







Amyloidosis is a heterogenous group of disorders that develops secondary to the deposition of abnormally folded proteins, amyloid fibrils, in the extracellular space. Amyloidosis is classified according to the type of the precursor protein that results in the formation of amyloid fibrils. It can be inherited or acquired, and can involve multiple organs, including the heart. Amyloidosis that involves the heart is referred to as cardiac amyloidosis (CA) in this review.
Epidemiology
Immunoglobulin light chain amyloidosis (AL amyloidosis) and transthyretin amyloidosis (ATTR amyloidosis) are the two most common forms of systemic amyloidosis. AL amyloidosis is a rare disease, with an incidence of approximately 9–15.2 cases per million person-years.1,2 However, autopsy studies have shown that acquired ATTR amyloidosis (formerly called senile cardiac amyloidosis) is not uncommon, affecting 10–25 % of people aged over 80 years, and 50 % of those over 90 years.3–5 The prevalence of ATTR amyloidosis is higher in the US than in Asia.5 CA has a higher prevalence in patients with severe aortic stenosis. Recent published data showed that 13.9–16 % of patients who undergo transcatheter aortic valve replacement have CA.6,7 The median survival of people with AL and ATTR amyloidosis is 211–330 days and 961–2,250 days, respectively.8,9
Pathophysiology
An amyloid fibril protein is defined as a protein that is deposited as insoluble fibrils, mainly in the extracellular spaces of organs and tissues as a result of sequential changes in protein folding resulting in a condition known as amyloidosis.10 In humans, there are 31 extracellular precursor proteins that can lead to amyloid fibril formation.10 Among those, the AL and ATTR amyloid proteins are those that traditionally involve the heart. AL is an amyloid protein derived from light chain immunoglobulin while ATTR is derived from misfolded unstable liver-based transthyretin protein. ATTR can be hereditary caused by a mutation in the ATTR gene; known as mutant ATTR (ATTRm). It can also be acquired; the acquired type is known as ATTR wild-type (ATTRwt) and used to be called senile cardiac amyloidosis.
Amyloid fibrils can be deposited in any structure in the heart. They can be found in the atria and ventricles, and involve the pericardium, myocardium and endocardium. The valves, the conduction system and the coronary vessels can be affected as well. Depositions vary from small nodules to complete replacement of the cardiac tissue.11 High-grade deposition(>50 % of the myocardium) and frequent (90 %) vascular involvement have been noted in hearts with AL amyloidosis compared with low-grade deposits and infrequent (4 %) vascular involvement in the ATTR type.12 Myocardial deposition can lead to stiff ventricles early in the course of the disease, resulting in diastolic dysfunction and usually followed by left ventricular systolic dysfunction.13,14
Clinical Manifestations
The impact of cardiac amyloid deposition can range from being asymptomatic to a rapidly fatal disease. CA tends to be evident clinically when the deposition exceeds 10 % of the myocardium.12 Early recognition can be important to reduce the disease burden and improve survival.
AL amyloidosis can affect any organ except the central nervous system leading to variety of symptoms. Outside the heart, it can result in the development of carpal tunnel syndrome, nephrotic syndrome, autonomic neuropathy, malabsorption syndromes, hepatomegaly, macroglossia (27.2 %), and periorbital purpura (12.5 %). Fatigue and weakness are the most common presenting symptoms. AL cardiac amyloidosis is unusual in isolation (3.9 % of patients).15
ATTRm can cause peripheral and autonomic neuropathy. Around 50 % of affected patients have a positive family history.16 ATTRwt can lead to carpal tunnel syndrome, biceps tendon rupture and spinal stenosis.17,18 It usually takes years before heart failure (HF) develops. AF and heart block can also develop (Tables 1 and 2).
Diagnosis
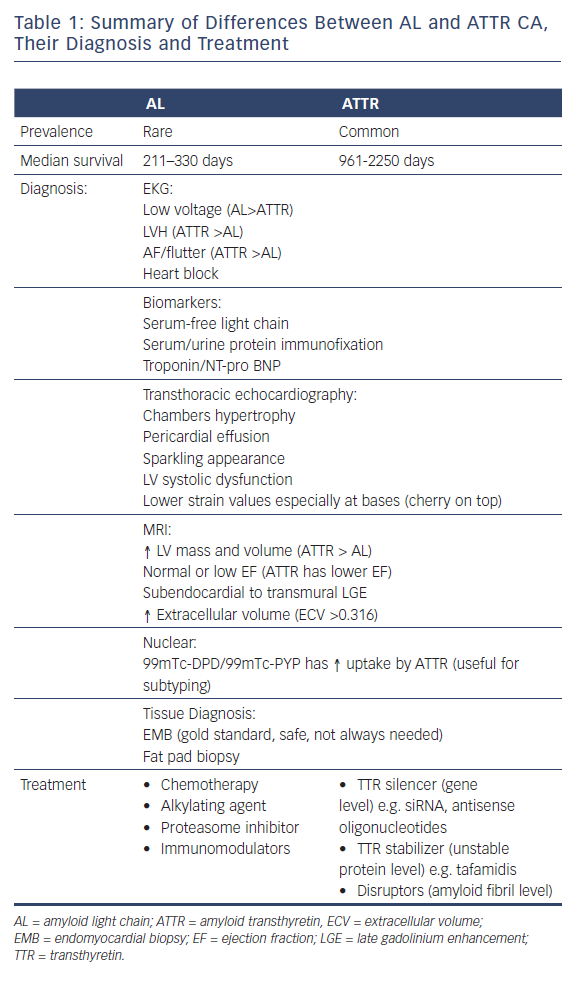
On EKG, around 46–70 % of CA cases have a low voltage (defined as all limb leads ≤0.5 mV, all precordial leads ≤1.0 mV, or Sokolow index ≤1.5 mV) at the time of diagnosis.8,19 Low voltage is more common in the AL than ATTR type (Figure 1).8,20 However, this difference between the subtypes is not consistent and has not been observed in other retrospective studies.21 Of patients with CA, 9–13 % meet the voltage criteria for left ventricular hypertrophy (LVH), and 15–42 % have atrial flutter or AF.8,21 Second degree atrioventricular block, LVH and AF tend to be more common in the ATTR than the AL subtype.8,21 QRS duration tends to be longer in the ATTR type.8
Biomarkers can be helpful in diagnosis and in determining the prognosis. Serum-free light chain and serum and urine protein immunofixation, which are more sensitive than serum and urine protein electrophoresis and have thus replaced this, are usually positive with AL amyloidosis and important for typing CA. Serum amyloid A, beta2-microglobulin, osteopontin, and osteoprotegerin are other markers that can be elevated early in the course of the disease, but have variable specificity for CA.22 Troponin elevation and elevated N-terminal pro B-type natriuretic peptide (NT-proBNP) levels are late markers and signify cardiac damage.23 NT-proBNP levels elevated out of proportion to left ventricular systolic dysfunction can be a sign of CA.24,25
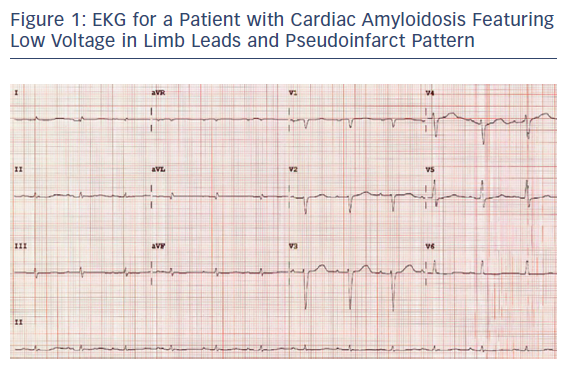
Imaging studies are helpful in the diagnosis of CA and may be sufficient to establish a diagnosis when combined with biomarker data in ATTR amyloidosis. Echocardiography is a useful screening tool, and findings for patients with proven CA include concentric left ventricular hypertrophy, small to normal left ventricular diameters (>90 %), left atrial enlargement (89–91 %), a granular and sparkling appearance of the myocardium (55–82 %), moderate to large pericardial effusion (64–67 %), and left ventricular systolic dysfunction (44–73 %) (Figure 2).14,20 The transmitral flow velocity is often compatible with abnormal relaxation in all types of CA, and is seen in 57–64 % of patients, usually in the early phase of the disease.13,14 Intracardiac thrombus has also been observed on echocardiograms of patients with AL CA.26
The majority of these findings, while more common in CA, are also seen in patients with hypertrophic cardiomyopathy (HCM). Right ventricular inferior wall thickening (>4 mm) has been seen in 25 % of patients with CA, compared to 15 % of those with HCM, and sparkling myocardium was present in 25 % of people with CA, compared to 12.5 % of those with HCM. Compared to HCM, CA leads to a more advanced stage of diastolic dysfunction and a more impaired ejection fraction.27 CA can also cause left ventricular outflow tract obstruction and be mistaken for hypertrophic obstructive cardiomyopathy. The 2D–speckle tracking strain values, especially at the bases, are consistently lower (i.e. more impaired) in people with CA than in those with HCM, which makes these deformation parameters better differentiating factors than traditional echocardiographic findings.27 The global longitudinal strain typically features preserved apical segments (cherry on the top) and reduced strain in the basal segments. The presence of pleural effusion is associated with a worse prognosis.
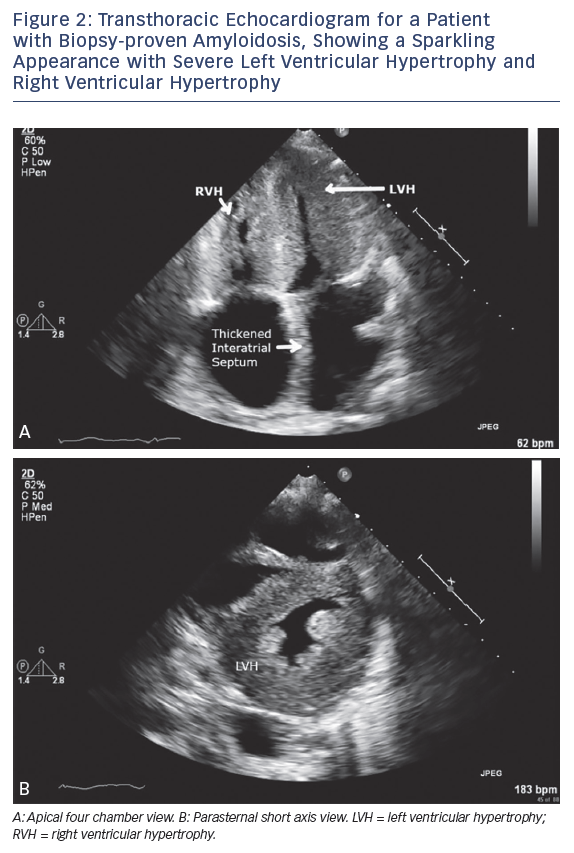
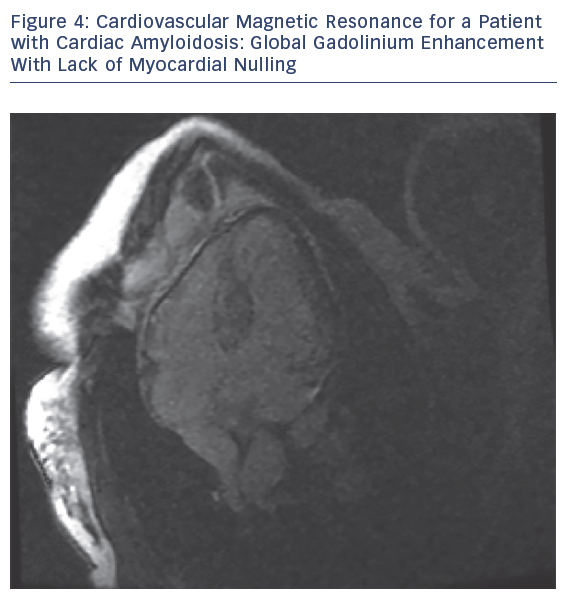
MRI is a valuable modality in evaluating CA. It helps by assessing biventricular function, mass and volume, late gadolinium enhancement (LGE), T1 mapping, extracellular volume (ECV) and strain pattern. Dungu et al. compared ATTR and AL types, and found that ATTR CA has greater LV mass and volume, lower EF, thicker interventricular septum, RV free wall and large atrial areas.28 LGE is thought be an early sign that may precede LV wall thickening (Figures 3 and 4).28
Maceria et al. found that 69 % of patients with CA have global subendocardial LGE with non-coronary distribution. LGE was present in all cases of ATTR CA and in the majority of those of AL CA.29 Dungu et al. and Fontana et al. showed that LGE in ATTR CA has more RV LGE, more transmural LGE and less global subendocardial LGE than AL CA.28,30 Based on these observations, an LGE-based scoring system was created to differentiate between ATTR and AL CA, and this system detected ATTR CA with an 87 % sensitivity and 96 % specificity.28 Furthermore, Barison et al. found that a cut‐off value of myocardial ECV >0.316 has a sensitivity of 79 % and a specificity of 97 % for differentiating between patients with amyloidosis and control subjects. Increased myocardial ECV was also correlated with disease severity.31 Cardiovascular magnetic resonance strain analysis can detect early systolic and diastolic strain impairment in patients with AL CA with and without LGE.32
The role of nuclear imaging in the typing of CA is increasing. Many radiotracers have been studied to help detect and diagnose CA with limited value. However, technetium-3,3-diphosphono-1,2-propanodicarboxylic acid (99mTc-DPD) and technetium pyrophosphate (99mTc–PYP) have shown great potential in differentiating between the two main types of CA. ATTR CA has high uptake to DPD and PYP in contrast to AL CA. A study by Bokhari et al. on 45 subjects with CA showed that the ATTR subtype has a significant higher myocardial tracer uptake than the AL subtype. Using a heart-to-contralateral ratio >1.5, consistent with intensely diffuse myocardial tracer retention, had a 97 % sensitivity and 100 % specificity for identifying ATTR CA.33 Another study on a smaller number of patients showed that 99mTc–DPD uptake, as measured by visual scoring, has 100 % sensitivity and specificity for ATTR CA.34 Various specialized CA centers have started to use this in combination with different biomarkers as a substitute for myocardial biopsy for diagnosing ATTR CA.

Tissue biopsy is the gold standard for diagnosing and subtyping amyloidosis. Endomyocardial biopsy (EMB) is usually carried out if there is a suspicion of CA and imaging modalities have not resulted in a diagnosis. It is a generally safe procedure if carried out by an experienced clinician. However, it is an invasive procedure and comes with considerable risks.35 In the right clinical settings and when at least four samples are obtained, EMB has a reported sensitivity of 100 % for detecting CA.36,37 Fat pad biopsy is a safer procedure that can obviate the need for EMB but has sensitivity of only 50–75 % in AL and 12–73 % in ATTR amyloidosis (Figure 5).38–40

Prognosis
AL CA is associated with a poorer prognosis than ATTR CA.21 Low voltage on EKG at the time of presentation is associated with all-cause mortality in AL and ATTR CA.8,21 On echocardiogram, left ventricular strain, an E/A ratio of >2.1, and a dt <150 ms are bad prognostic factors.41 On MRI, LGE was found to be a sign of poor prognosis in a study by Fontana et al. but this was an inconsistent finding.29 The APOLLO 3 study showed that NT-proBNP in ATTR amyloidosis is a strong predictor of survival.42 Patients with NT–proBNP levels of >3,000 ng/l have 19.3-fold greater mortality risk than those with levels below 3,000 ng/l. Other studies have shown NT–proBNP levels have a similar predictive value in AL amyloidosis.25
Management
Treatment of Amyloidosis
Treatment of amyloidosis depends on its type. AL amyloidosis is a plasma cell dyscrasia, and chemotherapy has been used to target the plasma cells with alkylating agents (e.g. cyclophosphamide), proteasome inhibitors (e.g. bortezomib), immunomodulators (e.g. pomalidomide), and anti-CD-38 monoclonal antibodies (IgG1-kappa, e.g. earatumumab). The goal of the therapy is to achieve hematological response, defined as the normalization of serum free light chain, and negative serum and urine monoclonal immunofixation. Triple chemotherapy with bortezomib, dexamethasone and an alkylating agent for AL CA has been shown to improve survival when hematological response was achieved.43,44 Daratumumab, reserved currently for relapsed or refractory cases, yielded rapid hematologic response in 76 % of patients with AL CA in a recent study.45 Monoclonal antibodies targeting amyloid fibrils in AL amyloidosis are in the phase of clinical trials and will hopefully provide a selective, targeted therapy for patients with AL CA.46
ATTR amyloidosis therapies are targeted at the gene (TTR silencers), the unstable protein (TTR stabilizers) and the amyloid fibril (disruptors). TTR silencers are either small interfering RNA (siRNA) or antisense oligonucleotides that target TTR messenger RNA in the hepatocyte, halting the production of TTR protein. Patisiran is a siRNA that was found to improve quality of life in patients with CA in the APOLLO 3 trial. In recent multicenter double-blind, placebo-controlled, clinical trial, Tafamidis, a TTR stabilizer showed improvement in all-cause mortality and quality of life with reduction in cardiovascular hospitalizations in patients with CA treated compared to placebo.47 Amyloid fibril disrupters, such as doxycycline with tauroursodeoxycholic acid, and green tea extracts, have been studied in small, open label studies, and showed no progression of wall thickness or decreased thickening of interventricular septal wall at 1 year.48,49
Treatment of Advanced Heart Block
If heart block develops, permanent pacemaker implantation should be considered.
Treatment of Heart Failure
Diuresis is the mainstay of HF management in patients with CA. This is usually a challenge, because there is commonly autonomic dysfunction associated with CA that may lead to hypotension. This complication can be managed with midodrine. Torsemide has greater bioavailability than furosemide and is therefore preferred because of malabsorption syndrome associated with CA,directly from AL amyloidosisor from gut edema from HF. Angiotensin-converting enzyme inhibitors are not tolerated, usually because of autonomic dysfunction. Calcium channel blockers can bind to amyloid fibrils, causing a greater negative inotropic effect. Digoxin can bind to amyloid fibrils but has no role in HF management. Beta-blockers tend to cause fatigue and worsen HF, so are usually avoided. Anticoagulation should be considered in patients with CA, especially those with the AL type, because of their increased risk of thrombosis.26
Conclusion
Cardiac amyloidosis, especially ATTR amyloidosis, is not as rare as previously thought. The advancement in cardiac imaging technologies, especially CMR, facilitated the early diagnosis of this disease and could potentially substitute the need for tissue diagnosis. With the recent development of effective life-prolonging therapies, there is compelling need to early diagnose and treat CA.