







Cardiovascular disease is an important cause of morbidity and mortality in patients with type 1 and especially type 2 diabetes mellitus.1,2 Data from prospective studies suggest that diabetes is associated with a two to fourfold excess risk of coronary heart disease and coronary death.3–5 It was therefore hoped that controlling hyperglycemia would reduce cardiovascular disease incidence and mortality, but unfortunately, landmark trials in type 2 diabetes have shown mixed results. In fact, the pendulum has swung in the opposite direction with concerns about the cardiac risks of rosiglitazone, culminating in the US Food and Drug Administration (FDA) 2008 guidelines requiring cardiovascular safety data in novel therapies for type 2 diabetes. In this article, we will review landmark trials in glycemic control, the rosiglitazone story, and post-2008 cardiovascular outcome studies of new type 2 diabetes medications.
Landmark Trials
The list of landmark studies in type 2 diabetes includes: the University Group Diabetes Program (UGDP), the United Kingdom Prospective Diabetes Study (UKPDS), Action to Control Cardiovascular Risk in Diabetes (ACCORD), Action in Diabetes and Vascular Disease: Preterax and Diamicron Modified Release Controlled Evaluation (ADVANCE), and the Veterans Affairs Diabetes Trial (VADT) (see Table 1). Following the UGDP and the UKPDS, there was little question that improved glycemic control would prevent the onset and delay the progression of microvascular disease. The main purpose of the ACCORD, the ADVANCE, and the VADT studies was to look at the effect of tight glycemic control on macrovascular outcomes in patients with type 2 diabetes.
Initiated in 1961, the UGDP was the first large, prospective, randomized trial designed to answer the question of whether control of blood glucose levels helps to prevent vascular disease in patients with non-insulin dependent diabetes. Up until that point, support for or against this hypothesis generally came in the form of expert opinion or from studies that suffered from being non-randomized and retrospective.6 The UGDP randomized 1,027 patients with recently diagnosed type 2 diabetes to one of five treatment groups:
- placebo;
- phenformin, a biguanide;
- tolbutamide, a sulfonylurea;
- fixed dose insulin; and
- variable dose insulin, with insulin dose adjusted to achieve a fasting blood glucose <110 mg/dl and a blood glucose <210 mg/dl one hour after ingestion of 50 grams of glucose.7–14
Median follow-up was more than 5 years. While all treatment groups achieved an initial reduction in fasting glucose, only the variable dose insulin group maintained this reduction in subsequent follow-up visits. Compared to placebo, none of the treatment groups demonstrated benefit in all-cause or cardiovascular mortality, and unexpectedly, tolbutamide and phenformin caused an increase in cardiovascular mortality. These findings led to early discontinuation of the tolbutamide and phenformin treatments.
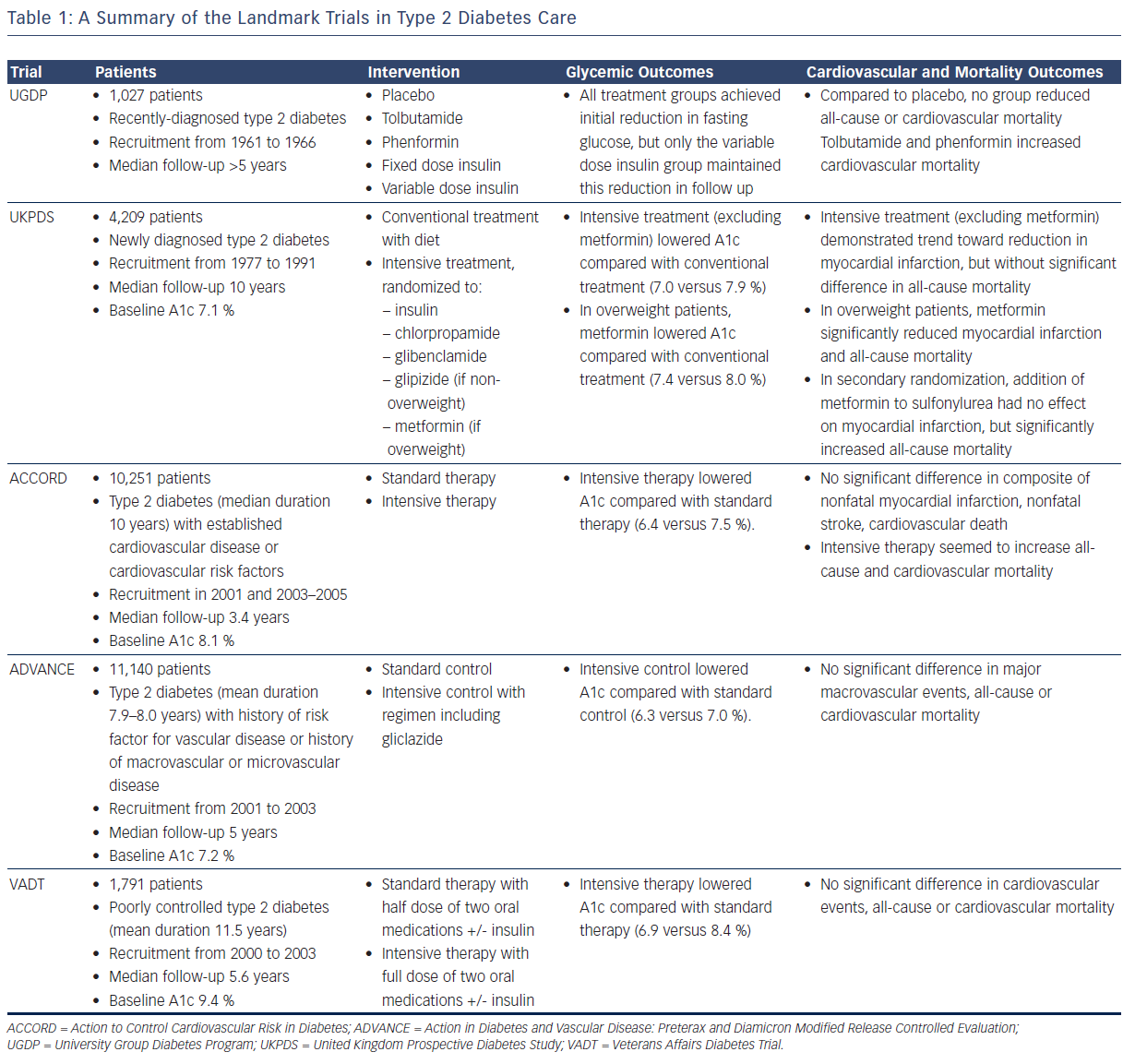
The UGDP reports provoked a great deal of controversy and criticism, largely because of the increased cardiovascular mortality with tolbutamide, and partly in response, the UKPDS was organized, again studying the morbidity and mortality impacts of insulin, sulfonylureas, and a biguanide, in this case metformin.15 In this study, 4,209 patients with newly diagnosed type 2 diabetes were recruited from 1977 to 1991.16,17 Average baseline hemoglobin A1c was 7.1 %. Non-overweight patients were randomized to conventional treatment with diet, or intensive treatment with insulin or a sulfonylurea (chlorpropamide, glibenclamide, or glipizide). Overweight patients were randomized to conventional treatment with diet, or intensive treatment with insulin or metformin or a sulfonylurea (chlorpropamide or glibenclamide). The goal of the conventional arm was to maintain fasting plasma glucose <270 mg/dl without hyperglycemia symptoms, while the goal of the intensive arm was fasting plasma glucose <108 mg/dl. Over 10 years of follow-up, the median A1c values were lower in the intensive group (excluding metformin) compared with the conventional group (7.0 versus 7.9 %). In a separate analysis of overweight patients, median A1c for the metformin group was 7.4 versus 8.0 % for conventional treatment. Excluding metformin, there was a trend toward reduction in myocardial infarction with intensive therapy (14.7 versus 17.4 events per 1,000 patient-years, P=0.052), but no significant difference in all-cause mortality. In overweight patients, metformin significantly reduced myocardial infarction (11.0 versus 18.0 events per 1,000 patient-years, P=0.01) and all-cause mortality (13.5 versus 20.6 events per 1,000 patient-years, P=0.011). Interestingly, a secondary randomization was performed in the UKPDS in which patients with inadequate glycemic control on sulfonylurea therapy were randomized to addition or no addition of metformin. A1c over 4 years of follow-up decreased with addition of metformin (7.7 versus 8.2 %), but there was no significant difference in myocardial infarction and actually an increase in all-cause mortality (30.3 versus 19.1 events per 1,000 patient-years, P=0.041). It is important to point out that these latter results did not affect clinical practice in terms of adding metformin to a sulfonylurea, and that statistical flukes are possible even with large multicenter trials.
The UGDP and the UKPDS came to different conclusions regarding sulfonylureas, and to this date, it is still unclear what effect this class of medications has on mortality. A recent 2013 review of 72 randomized controlled trials concluded that there was insufficient evidence to determine whether sulfonylureas increase all-cause or cardiovascular mortality.18 In some ways, however, the concern regarding cardiovascular harm with sulfonylurea therapy overshadowed the fact that neither the UGDP nor the UKPDS showed significant cardiovascular benefit with intensive insulin therapy.19 In addition, the primary metformin randomization in the UKPDS led to a very different result compared with the secondary metformin randomization. In the wake of these conflicting results, three large trials, the ACCORD, the ADVANCE, and the VADT, set out to answer the still-unresolved question of whether intensive glucose control in type 2 diabetes reduces cardiovascular complications.19–21
In the ACCORD trial, 10,251 patients with existing type 2 diabetes (median duration 10 years) and either established cardiovascular disease or additional cardiovascular risk factors were recruited in two phases, in 2001 and also from 2003 to 2005.22 Median baseline A1c was 8.1 %. Patients were randomized to standard therapy targeting an A1c of 7.0–7.9 %, or intensive therapy targeting an A1c <6.0 %. The primary outcome was a composite of nonfatal myocardial infarction, nonfatal stroke, or death from cardiovascular causes. Median A1c was lower in the intensive group (6.4 versus 7.5 %). No significant difference was seen in the primary outcome, but intensive therapy seemed to increase all-cause mortality (5.0 versus 4.0 %, P=0.04) and cardiovascular mortality (2.6 versus 1.8 %, P=0.02). These results led the study’s external data safety monitoring board to end the trial early, and prompted a worldwide discussion on what the glycemic goals should be in type 2 diabetes, especially for older patients with a history of coronary heart disease.
In the ADVANCE study, 11,140 patients with existing type 2 diabetes (mean duration 7.9–8.0 years) and a history of macrovascular disease or microvascular disease or risk factor for vascular disease were recruited from 2001 to 2003.23 Median baseline A1c was 7.2 %, and median follow-up was 5 years. Patients were randomized to intensive control, targeting an A1c of 6.5 % or less with therapy including gliclazide modified release, or standard control based on local guidelines. The primary outcomes were composites of macrovascular events (nonfatal myocardial infarction, nonfatal stroke, or cardiovascular death) and microvascular events (new or worsening nephropathy or retinopathy). At the end of follow-up, the intensive group achieved a lower median A1c (6.3 versus 7.0 %). Intensive control also reduced the incidence of combined major macrovascular and microvascular events (18.1 versus 20.0 %, P=0.01), primarily due to a reduction in nephropathy. There was no significant difference in major macrovascular events, cardiovascular mortality, or all-cause mortality.
In the VADT, 1,791 patients with poorly controlled type 2 diabetes (mean duration 11.5 years) were recruited from 2000 to 2003.24 Mean baseline A1c was 9.4 %, and median follow-up was 5.6 years. Patients were randomized to standard therapy (half-maximal dose of two oral medications with insulin added if A1c was not <9 %) or intensive therapy (maximal dose of two oral medications with insulin added if A1c was not <6 %). The two oral medications were chosen based on baseline body mass index (BMI): patients with BMI 27 kg/m2 or more were started on metformin plus rosiglitazone, while patients with BMI <27 kg/m2 received glimepiride plus rosiglitazone. The primary composite outcome was time to first occurrence of any cardiovascular event (myocardial infarction, stroke, cardiovascular death, new or worsening congestive heart failure, amputation for ischemic gangrene, inoperable coronary artery disease, or surgical intervention for cardiac, cerebrovascular, or peripheral vascular disease). The intensive group achieved a lower A1c (6.9 versus 8.4 %). There was no significant difference in the primary composite outcome, or in all-cause or cardiovascular mortality.
Two different groups of patients were studied in these landmark trials, with the UGDP and the UKPDS studying recently diagnosed patients (although many of these subjects most likely had undiagnosed diabetes for many years), and the ACCORD, the ADVANCE, and the VADT studying patients with long-standing diabetes of 8–12 years duration and with documented cardiovascular disease or risk factors for cardiovascular disease. The glycemic control achieved in the UGDP is difficult to compare to that of the UKDPS as no A1c values were reported in the former, primarily because the utility of glycated hemoglobin had not been demonstrated prior to the development of the UGDP protocol. However, in many ways, the UKPDS served as a rebuttal to the UGDP findings of increased cardiovascular mortality with tolbutamide and phenformin. The UKPDS reported a trend toward reduced myocardial infarction and no mortality difference with intensive therapy with insulin or sulfonylurea. It also reported a statistically significant benefit in myocardial infarction and all-cause mortality with metformin in overweight newly-diagnosed type 2 diabetic patients. It is important to acknowledge that this positive result is based on a relatively small number of patients, 342 in the metformin arm. In addition, the secondary randomization protocol produced the confusing result of increased all-cause mortality and a lack of benefit in myocardial infarction when metformin was added to sulfonylurea therapy.
Both the ACCORD and the ADVANCE trials studied large numbers of patients with long-standing type 2 diabetes, but produced different results in terms of cardiovascular and all-cause mortality. One difference between the two studies was baseline A1c, with the ACCORD trial patients having a higher baseline A1c, indicating worse control leading up to study enrollment. Also, the A1c difference between the intensive and standard groups was wider in the ACCORD study, possibly allowing a mortality difference to be detected between the two treatment groups. On the other hand, patients in the VADT had even worse baseline glycemic control prior to enrollment, and an even wider gap in A1c between the treatment groups, but their results did not echo the ACCORD study results.
In summary, despite several large trials over the years, intensive glycemic control of type 2 diabetes has not convincingly improved macrovascular outcomes. The general consensus at this time is that the effect of glucose control on cardiovascular disease in type 2 diabetes, if it exists, is minimal.
The Rosiglitazone Story
In the setting of uncertainty about the cardiovascular benefit of glycemic control, controversy developed over the potential cardiovascular harms of rosiglitazone, over and above the well-accepted effect of thiazolidinediones on heart failure. In 2003, the World Health Organization reported a signal for cardiac disease and thiazolidinediones from a database of adverse reaction reports. Following this, GlaxoSmithKline began analyzing phase II and III randomized controlled trials of rosiglitazone for excess cardiovascular risk. This meta-analysis was presented to the FDA in August 2006, with the conclusion that there was an increase in cardiac ischemic events with rosiglitazone (hazard ratio 1.31, 95 % confidence interval 1.01–1.70).25 In May 2007, Nissen and Wolski published a separate meta-analysis that found a significant increase in myocardial infarction with rosiglitazone (odds ratio 1.43, P=0.03) and a trend toward increased cardiovascular mortality with rosiglitazone (odds ratio 1.64, P=0.06).26 The latter study garnered a great deal of press and even Congressional attention, although it also received criticism that highlighted the difficulty in pooling data from disparate trials to draw conclusions about relatively rare adverse events. For example, it was noted that many of the included trials were of short duration and not designed to evaluate cardiovascular outcomes, there was heterogeneity in patient populations and drug dosing regimens studied, and trials were excluded if they reported no myocardial infarction or cardiovascular death events.27 In 2009, results of the Rosiglitazone Evaluated for Cardiovascular Outcomes in Oral Agent Combination Therapy for Type 2 Diabetes (RECORD) trial were reported.28 This was a large randomized prospective trial of 4,447 patients looking at cardiovascular outcomes after addition of rosiglitazone to either metformin or sulfonylurea, compared to the combination of metformin and sulfonylurea. After a mean follow-up of 5.5 years, there was no significant difference in the primary composite outcome of cardiovascular hospitalization or cardiovascular death. Due to allegations of data mishandling, the FDA required an independent re-adjudication of these results. This was performed by the Duke Clinical Research Institute and presented to the FDA in June 2013, confirming the results of the original RECORD publication in showing no difference in cardiovascular outcomes. The FDA subsequently removed its prescribing and dispensing restrictions for rosiglitazone in November 2013, and fully eliminated its rosiglitazone Risk Evaluation and Mitigation Strategy in December 2015.
It is worth noting that pioglitazone, another thiazolidinedione, was the subject of the Prospective Pioglitazone Clinical Trial in Macrovascular Events (PROactive), a randomized trial of 5,238 patients.29 This study reported no significant difference in its primary composite endpoint (all-cause mortality, nonfatal myocardial infarction, stroke, acute coronary syndrome, surgical intervention in the coronary or leg arteries, or amputation above the ankle) but did report significant benefit with pioglitazone in the main secondary composite endpoint of all-cause mortality, nonfatal myocardial infarction, and stroke.
New Therapies
In part due to the rosiglitazone controversy, the FDA issued guidelines in December 2008 on the evaluation of cardiovascular risk in the development of new therapies for type 2 diabetes. In essence, unless a phase III study demonstrates cardiovascular superiority (upper bound of the 95 % confidence interval of the risk ratio <1.0) or non-inferiority (upper bound of the 95 % confidence interval of the risk ratio <1.3) of a new therapy compared with placebo, a postmarketing safety trial will generally be necessary.30 Thus far, six trials have been published, looking at saxagliptin, alogliptin, sitagliptin, lixisenatide, liraglutide, and empagliflozin, respectively, with many more to come (see Table 2).
Saxagliptin, alogliptin, and sitagliptin are inhibitors of dipeptidyl peptidase 4 (DPP-4), decreasing the degradation of endogenous glucagon-like peptide 1 (GLP-1) by DPP-4 and thereby enhancing pancreatic insulin secretion and inhibiting glucagon release. Saxagliptin was studied in the Saxagliptin Assessment of Vascular Outcomes Recorded in Patients with Diabetes Mellitus – Thrombolysis in Myocardial Infarction (SAVOR-TIMI 53) trial, which randomized 16,492 patients with type 2 diabetes and cardiovascular risk factors or history of cardiovascular events to receive saxagliptin or placebo.31 There was no significant difference in the primary composite endpoint of cardiovascular death, nonfatal myocardial infarction, or nonfatal ischemic stroke (hazard ratio 1.00, P<0.001 for non-inferiority). However, there was an increased risk of hospitalization for heart failure with saxagliptin (hazard ratio 1.27, P=0.007). Alogliptin was studied in the Examination of Cardiovascular Outcomes with Alogliptin Versus Standard of Care (EXAMINE) trial, which randomized 5,380 patients with type 2 diabetes and recent myocardial infarction or hospitalization for unstable angina to receive alogliptin or placebo.32 There was no significant difference in the primary composite endpoint of cardiovascular death, nonfatal myocardial infarction, or nonfatal stroke (hazard ratio 0.96, P<0.001 for non-inferiority). Given the saxagliptin result concerning heart failure, an exploratory analysis of the EXAMINE trial was undertaken, which did not show any increased risk of heart failure with alogliptin.33 Finally, sitagliptin was studied in the Trial Evaluating Cardiovascular Outcomes with Sitagliptin (TECOS) trial, which randomized 14,671 patients with type 2 diabetes and established cardiovascular disease to receive sitagliptin or placebo.34 There was no significant difference in the primary composite endpoint of cardiovascular death, nonfatal myocardial infarction, nonfatal stroke, or hospitalization for unstable angina (hazard ratio 0.98, P<0.001 for non-inferiority), and no significant difference in the rate of hospitalization for heart failure (hazard ratio 1.00, P=0.98).
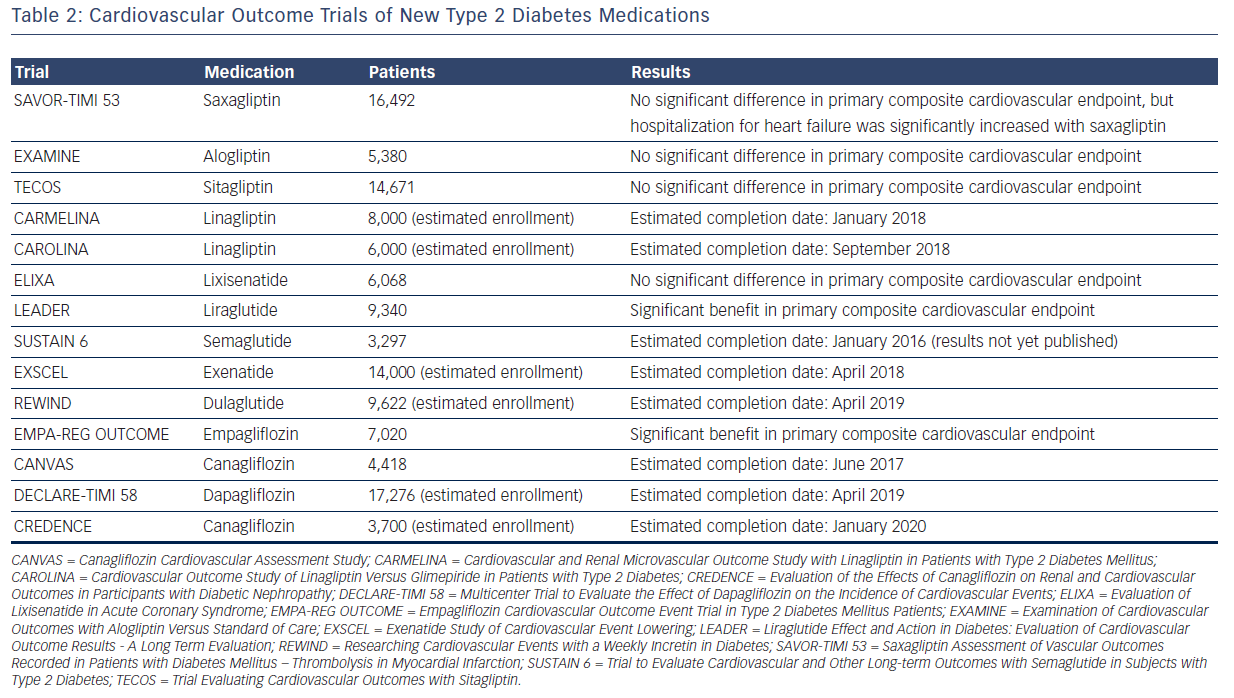
Lixisenatide and liraglutide are in the class of diabetes medications that directly activate the receptor for endogenous GLP-1. Lixisenatide was studied in the Evaluation of Lixisenatide in Acute Coronary Syndrome (ELIXA) trial, which randomized 6,068 patients with type 2 diabetes and recent acute coronary event to receive lixisenatide or placebo.35 There was no significant difference in the primary composite endpoint of cardiovascular death, nonfatal myocardial infarction, nonfatal stroke, or hospitalization for unstable angina (hazard ratio 1.02, P<0.001 for non-inferiority), and no significant difference in hospitalization for heart failure (hazard ratio 0.96, P=0.75). The results of liraglutide's cardiovascular safety trial were recently presented at the 2016 meeting of the American Diabetes Association and simultaneously published online in the New England Journal of Medicine.36 In the Liraglutide Effect and Action in Diabetes: Evaluation of Cardiovascular Outcome Results (LEADER) trial, 9,340 patients with type 2 diabetes at high risk for cardiovascular disease were randomized to receive liraglutide or placebo. The primary composite outcome (death from cardiovascular causes, nonfatal myocardial infarction, or nonfatal stroke) occurred in fewer patients in the liraglutide group than in the placebo group (hazard ratio 0.87, P=0.01 for superiority). In addition, there was a significant benefit with liraglutide in the exploratory outcomes of cardiovascular mortality (hazard ratio 0.78, P=0.007) and all-cause mortality (hazard ratio 0.85, P=0.02). ELIXA and LEADER trials led to different results; however, it is not clear if these were due to differences in the study design, demographics of the subjects, or inherent differences between lixisenatide and liraglutide.
Empagliflozin is an inhibitor of sodium-glucose cotransporter 2 (SGLT-2), leading to decreased renal glucose reabsorption and increased urinary glucose excretion. In the Empagliflozin Cardiovascular Outcome Event Trial in Type 2 Diabetes Mellitus Patients (EMPA-REG OUTCOME), 7,020 patients with type 2 diabetes at high risk for cardiovascular events were randomized to empagliflozin or placebo.37 Empagliflozin demonstrated a significant benefit in the primary composite endpoint of cardiovascular death, nonfatal myocardial infarction, or nonfatal stroke (hazard ratio 0.86, P=0.04 for superiority). In terms of secondary outcomes, there was less cardiovascular mortality (hazard ratio 0.62, P<0.001), all-cause mortality (hazard ratio 0.68, P<0.001), and hospitalization for heart failure (hazard ratio 0.65, P=0.002) with empagliflozin; although there was a non-significant increase in silent myocardial infarction and stroke. The mechanisms behind these cardiovascular benefits, which seemed to appear early in the study, remain speculative, and may include effects on blood pressure, weight, the renin-angiotensin system, and volume status. Additionally, further research is needed to determine if this is a class effect shared by all SGLT-2 inhibitors, and if benefit is limited to secondary cardiovascular prevention or extends to primary prevention as well.
Clinical Care
Metformin is considered first-line therapy for type 2 diabetes,38,39 and particularly in resource-limited practice settings, sulfonylureas are often still used when dual therapy is needed. Increasingly, however, practitioners have been turning to newer medications as second-line agents, including the DPP-4 inhibitors, GLP-1 receptor agonists, and SGLT-2 inhibitors, which have advantages in reducing hypoglycemia risk and weight gain. On top of these advantages, cardiovascular benefit has now been demonstrated with empagliflozin and liraglutide, which may lead to prioritization of these medications within type 2 diabetes treatment algorithms for patients at high cardiovascular risk.
Conclusion
While intensive glycemic control in type 2 diabetes has reduced microvascular complications, the expected improvement in cardiovascular outcomes has not clearly materialized in large randomized studies. In fact, intensive control may worsen all-cause and cardiovascular mortality in patients with a long history of poorly controlled type 2 diabetes at high risk for cardiovascular disease.
Disturbingly, concerns about rosiglitazone’s safety from GlaxoSmithKline’s presentation to the FDA and Nissen and Wolski’s 2007 publication led to the requirement that all new diabetes medications have to be evaluated in long-term cardiovascular outcome trials. However, to date, none of the subsequent cardiovascular studies of new diabetes therapies have demonstrated harm, except for an increase in heart failure hospitalizations with saxagliptin that has not been seen with other drugs in the same class. Given the long duration and high cost of these studies, typically measured in the hundreds of millions of dollars, many leaders in the field feel strongly that this requirement should be abolished as it is hampering diabetes drug development.
Finally, the SGLT-2 inhibitor empagliflozin and GLP-1 receptor agonist liraglutide have produced a great deal of excitement in the diabetes community after showing a significant cardiovascular benefit in patients with established cardiovascular disease. It remains to be seen where this excitement will lead and what future studies will reveal.