










MI was once considered a major health concern in industrialized nations, but is now seen routinely worldwide including in developing countries, which bear the greatest burden of cardiovascular disease.1 Coronary artery thrombosis is the final pathologic finding in most cases of MI and is mainly caused by atherosclerotic coronary artery disease (CAD).2 Our understanding of ‘vulnerable plaques’, defined as rupture-prone (or event-prone) plaques, has advanced dramatically in the last two decades, along with its recognition and subsequent treatment. This has been the result of advancements in basic and clinical investigations along with the development of imaging techniques and treatment technologies. However, at the same time, the results of vulnerable-plaque-oriented approaches to accomplish personalized risk assessment, stratification and treatment have room for significant improvement. In this review, we discuss the recent advancements in our understanding of vulnerable plaque and its management strategies from pathology to clinical management.
Plaque Phenotype as a Cause of Coronary Thrombosis
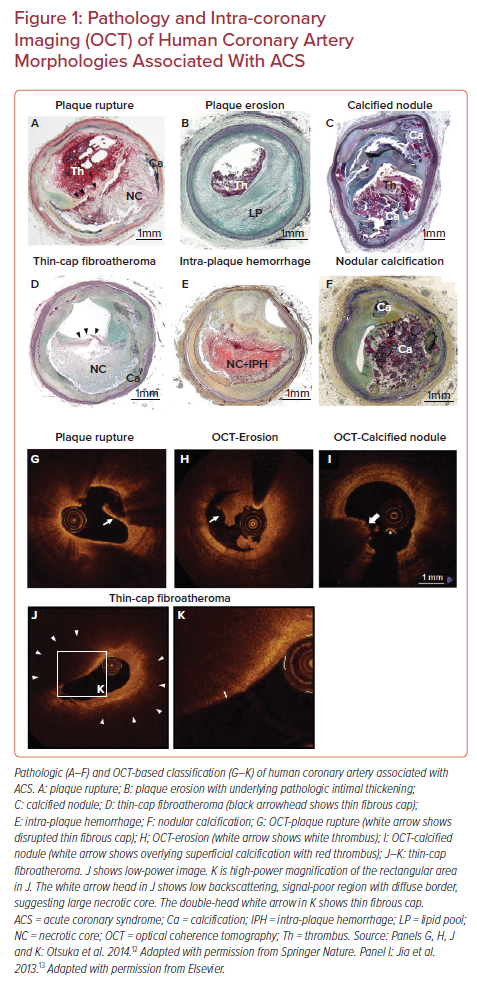
Prior autopsy studies have recognized three distinct morphologic entities leading to coronary thrombosis: plaque rupture (PR), plaque erosion (PE), and calcified nodules (CN; Figures 1A–1C).3 Our autopsy series of more than 800 people who died suddenly because of coronary artery thrombosis revealed that 55–60% of cases had underlying PR, 30–35% of the etiology was PE, and in 2–7% it was because of CN.2 A worldwide review of 22 autopsy studies, including 1,847 cases of hospital-based acute MI and sudden coronary death, also showed that 73% of fatal coronary thrombi originated from PR and 27% from PE.4
Plaque Rupture
PR refers to an advanced atheromatous lesion consisting of necrotic core with an overlying ruptured thin fibrous cap. ‘Rupture’ represents a structural defect in the fibrous cap that separates the highly thrombotic necrotic core contents from the bloodstream.5 It has been considered that the site of rupture is usually located at its mechanically weakest point, often near the shoulder regions.6 However, this is not always the case, as we have observed a comparable number of ruptures at the mid portion of the fibrous cap, especially in individuals who are dying during exertion.7 Therefore, it is reasonable that multiple processes can be involved in the mechanisms of fibrous cap rupture, e.g. fibrous cap degradation by matrix metalloproteinases released from inflammatory cells, great wall shear stress, and macrophage and smooth-muscle-cell apoptosis in the cap, as previously reported.8–10 The exposure to the blood of the lipid-rich necrotic core containing a large amount of pro-thrombotic tissue factor activates the coagulation cascade, resulting in the occlusion of the coronary artery by thrombus formation and subsequent MI.11 As will be discussed below, the vulnerable plaque paradigm is largely based on the mechanisms and morphologic characteristics thought to promote PR.
Plaque Erosion
The second common cause of intracoronary thrombosis is PE, which is defined as intracoronary thrombosis without evidence of fibrous cap rupture but that may include a necrotic core.3 As pathologic studies have revealed, the denudated endothelial layer is usually observed in the culprit site of PE.3 Exposure of underlying proteoglycans and collagen could be the nidus for thrombus formation. Generally, the underlying plaque phenotype is fibroatheroma with a thick fibrous cap or pathologic intimal thickening. Recent innovation of in vivo intra-coronary imaging modalities, such as optical coherence tomography (OCT), has allowed us to understand the underlying etiology of acute coronary syndromes (ACS) in the clinical setting.12 OCT studies of clinically determined ACS cases suggested the prevalence of PE is 27–31%.13,14 According to pathology and OCT studies, individuals of younger age (i.e. <50 years), female sex, and smokers are more likely to develop PE compared to PR.13,15,16 Moreover, traditional risk factors (e.g. diabetes, hypertension, hyperlipidemia, and chronic kidney disease) are less common in PE than PR.17,18 Local disturbed blood flow, toll-like receptor-2 activation in endothelial cells, endothelial apoptosis, concentrated extracellular matrix (e.g. hyaluronan and versican) in the subendothelial intimal layer, granulocyte rich inflammatory response, and neutrophil extracellular trap formation are considered to be underlying mechanisms of PE.19 However, the precursor lesion to thrombotic PE is still unclear, and the exact series of events that predispose to PE are unknown. Therefore, at present, the concept of vulnerable plaque pertaining to PE has not been developed and requires further investigation.
Calcified Nodule
CN is a third common mechanism of intracoronary thrombosis, with a prevalence of 3.3% in our autopsy case series who died suddenly due to acute coronary thrombosis.3 OCT-based clinical studies suggest the prevalence of CN in ACS is 2–8%.13,20 Pathologically, CN is defined as a disrupted luminal surface by nodules of dense calcium with overlying thrombus and little or no underlying necrotic core in arteries that are highly calcified, tortuous, and often have large sheets of calcification. Pathologic definitions of ‘eruptive calcified nodule’ (Figure 1C) and ‘nodular calcification’ (Figure 1F) need to be differentiated; the latter occurs within the plaque and does not involve disruption of the fibrous cap or contact with the lumen, but it is often associated with medial wall disruption with or without extension into the adventitia. The mechanism of fibrous cap disruption in CN causing overlying thrombosis is thought to be the fragmentation of necrotic core calcification by mechanical means. Areas of CN are often sandwiched between proximal and distal hard sheet calcification (most frequent in the mid-right coronary artery or left anterior descending artery-sites of maximal torsion).3,21 These sites of nodular calcification serve as sites of hinge motion and repeated trauma is thought to encourage the creation of nodules of calcium that can protrude into the lumen causing thrombosis. However, in order to define the precursor lesion to CN, further clinical investigations with better resolution imaging modalities need to be conducted.
What is the Concept of Vulnerable Plaque?
Thin-cap Fibroatheroma as a Precursor Lesion of Plaque Rupture
In the 1980s, James Muller coined the term ‘vulnerable plaques’ as precursor lesions from which MIs frequently developed.22 The morphological criteria for the definition of thin-cap fibroatheroma (TCFA) or vulnerable plaque originated from the concept that lesions that precede PR should have similar features (Figure 1D). Generally, ruptured plaques have large necrotic cores with ruptured thin fibrous caps accompanied by inflammatory cell infiltration including macrophages and – to a lesser extent – T lymphocytes, along with scant or a complete lack of smooth muscle cells. Our pathologic observation in cases of coronary PR revealed the thickness of the fibrous cap close to the rupture site was 23 ± 19 μm, with 95% of caps measuring <65 μm.23 Lesions with intact (non-ruptured) fibrous caps of <65 μm are also detected in non-culprit sites/vessels in patients dying of acute PR and are designated vulnerable plaques or TCFAs. In the year 2000, TCFA as the vulnerable plaque phenotype was first highlighted in a modified American Heart Association consensus described by Virmani et al.3 Subsequently, TCFA as the precursor lesion of PR became widely recognized; therefore, the original definition of vulnerable plaque is exclusively applicable to the PR paradigm.
Similar to PR, most TCFAs locate in the proximal segment of major coronary arteries.24 The pathologic features of TCFA are different from ruptured plaques in terms of smaller necrotic cores, less macrophage infiltration in the fibrous cap, and less calcification.25,26 Additionally, 70% of cases of sudden coronary death by acute PR commonly show evidence of TCFAs in non-culprit lesion or vessels.27 The frequency of TCFA was much smaller (around 30%) in people who died with flow-limiting severe coronary stenosis with stable fibrocalcific plaques. These findings imply that the presence of TCFA and rupture are not directly correlated (i.e. some TCFA may proceed to rupture while others do not).26 In addition, previous basic studies have revealed that inflammation, matrix metalloproteinases activity, and necrotic core expansion are all enhanced when TCFA is transforming into unstable plaques.8,28–32 However, precise triggers critical to the phenotypic transformation of TCFA plaques are still unclear.
Intra-Plaque Hemorrhage: Another Vulnerable Plaque Phenotype
Intra-plaque hemorrhage (IPH; Figure 1E) is considered one of the factors contributing to plaque destabilization and sudden increase in plaque volume.32,33 In 1987, Glagov reported that atherosclerotic coronary arteries undergo a progressive enlargement (i.e. positive remodeling) that allows preservation of the lumen area up to a point.34 Stenosis of the lumen only occurs once a plaque develops beyond 40% cross-sectional area (CSA) narrowing.34 Repeated subclinical PR and healing with luminal thrombus, as well as IPH (without luminal thrombus), are two major critical contributors for progressive plaque expansion until vessel occlusion and symptom onset.35 According to a large series of human plaques from cases of sudden coronary death, signs of previous IPH were frequently found in high-risk plaques prone to rupture compared to early lesion and stable plaques. Previously, our group showed the accumulation of erythrocyte membranes that contain a large amount of free cholesterol at the site of IPH contributes to the necrotic core expansion and further plaque vulnerability.36 Moreover, IPH is the essential trigger for macrophage phenotypic conversion into alternative subtypes distinct from lipid-laden foamy macrophages.37 IPH results in erythrocyte lysis through oxidative stress with release of free hemoglobin.38 Free hemoglobin immediately forms complexes with plasma haptoglobin, and macrophages internalize hemoglobin–haptoglobin complexes via the CD163 scavenger receptor for effective clearance.39,40 Recently, we reported that the mechanisms of CD163-positive alternative macrophages provoke micro angiogenesis and microvascular permeability by releasing vascular endothelial growth factor responding to hemoglobin–haptoglobin stimuli.41 Thereby, small amounts of intimal angiogenesis and bleeding could be exacerbated by the inflammatory response, initiating a vicious cycle whereby bleeding begets more bleeding. Collectively, both components, including cholesterol-rich erythrocyte membrane and released hemoglobin at the site of IPH, may synergistically accelerate plaque vulnerability leading to further adverse cardiovascular complications.
Calcification in the Plaque: The Contradictory Role on Plaque Vulnerability
Calcification is one of the common features of advanced atherosclerotic lesions, which develops and progresses with plaque type and luminal narrowing.42 A robust amount of clinical data has confirmed that the degree of coronary artery calcification is directly related to adverse cardiovascular outcomes in all populations, and it is a more reliable marker for future events than using risk equations based upon traditional risk factors (e.g. Framingham risk index).43 However, whether the coronary calcified plaques directly cause cardiac events or are just a reliable surrogate marker for the presence of CAD burden on a population-based level remains uncertain. Indeed, whether the presence of coronary calcification predicts plaque instability or stability is a crucial question for daily practice. One cannot treat coronary artery calcification as an all-or-none variable.
The type of calcification, location, volume, as well as density may all affect clinical risk and outcomes in distinctive ways.44 A study conducted detailed pathologic analysis of calcification, which included 510 coronary segments (17 cases) from acute MI cases, as well as 450 segments (15 cases) from age-matched controls who died of non-cardiac causes.45 In patient-based analysis, calcification was more abundant in cases of acute MI versus controls. However, in lesion-based analysis, an inverse correlation was observed between the extension of calcification and cap inflammation. Multivariate regression analysis confirmed that the calcification was not correlated with the presence of unstable plaques.45 Recent imaging-based human studies suggested that spotty calcification predicts plaque vulnerability in patients presenting with ACS, while large and heavy calcification correlates with overall plaque burden in the coronary tree.46 Spotty calcification with positive vessel remodeling and low attenuation, detected by coronary CT angiography (CCTA), was more frequent in patients with ACS or likely to develop ACS in the short term.47 This imaging-based spotty calcification (<3 mm diameter) is comparable with pathological fragmented calcification, which is frequently found in the outer rim of necrotic core (this should not be confused with microcalcification mentioned below).48 According to these pathologic observations, progressive calcification of human plaque occurs from the outer rim of the necrotic core into the surrounding collagenous matrix while the central core is preserved at this stage.48 When the calcification progresses into the central necrotic core, the calcification turns into sheets, a typical feature of stable calcified plaque. Therefore, in pathology, coronary calcium burden is greater in stable fibrocalcific plaques than unstable plaques including PR and TCFA and exhibit an opposite correlation with necrotic core area. Coronary calcium scoring might be helpful to detect the general risk of adverse coronary events in a population, i.e. suggesting the presence of coronary atherosclerotic plaque. However, it is not useful specifically to prospectively identify a culprit lesion of a future ACS.
Regarding direct correlation between calcification and plaque vulnerability, Vengrenyuk et al. proposed the role of microcalcification in the cap as one of the contributors of fibrous cap rupture behaving as local tissues stress concentrators.49,50 However, pathologically defined microcalcification (i.e. 0.5–15 µm diameter) requires extremely high spatial resolution to visualize and cannot be evaluated by current in-vivo clinical imaging.49–52 The detection of microcalcification in the fibrous cap could potentially be one of the predictors for vulnerable plaque. However, innovations in coronary imaging modalities are necessary to make them useful for this purpose.
Identification of Vulnerable Plaque by Imaging Modalities
A comprehensive risk stratification with high accuracy for determining an individual’s cardiovascular risk is desired. The scoring tools based on traditional risk factors such as age, sex, smoking, blood pressure, diabetes, and cholesterol levels (e.g. Framingham risk score, the Systematic Coronary Risk Evaluation, and Atherosclerotic Cardiovascular Disease risk estimator) could successfully stratify cardiovascular risk in whole populations.43,53,54 However, their effectiveness is limited by variations in lifetime risk level of patients and/or discrepancies between different algorithms arising from varying risk factors evaluated. Thus, in general practice, the use of currently developed risk stratification algorithms is low because of their oversimplification and concerns with over prescribing of medications.55 Moreover, most cardiovascular events occur in patients classified as low or intermediate risk by traditional risk factors.56
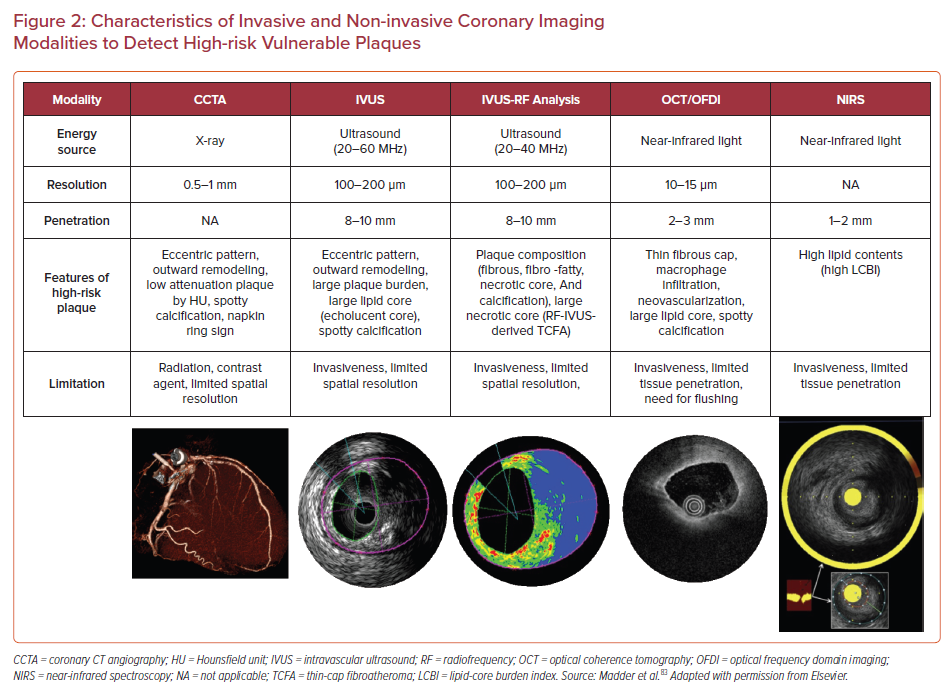
Therefore, more reliable atherosclerotic lesion-based stratifications by coronary imaging modalities are still considered worthy of study by cardiovascular clinicians and researchers. The characteristics of imaging modalities for high-risk coronary plaque detection and the summary of large clinical trials examining the effectiveness for these modalities are shown in Figure 2 and Table 1.57–69
Coronary CT Angiography
Substantial evidence suggests that coronary plaque burden is associated with the likelihood of future cardiovascular events. Coronary artery calcium score as assessed by CCTA has been shown to be predictive of coronary events in different ethnic populations independently of standard risk factors or scores.70 Further, an assessment of plaque vulnerability by CCTA is also thought to improve the diagnostic accuracy for patients with CAD. CCTA can provide reliable assessment in terms of the presence, size, and thickness of necrotic core, by grading tissue in Hounsfield units (HU).71 Currently accepted high-risk plaque characteristics by CCTA include positive remodeling (remodeling index ≥1.1), low-attenuation (a focal central area of plaque with an attenuation density of <30 HU), spotty calcification (<3 mm in maximum diameter), and napkin-ring sign (i.e. a central area of low-attenuation plaque with a peripheral rim of high attenuation). These high-risk features are robust markers of rupture-prone lesions supported by evidence gained from multiple clinical studies. For instance, the recently reported SCOT-HEART trial, which included 1,769 cases with 5 years of follow-up, demonstrated that patients with adverse plaque characteristics (i.e. positive remodeling or low attenuation plaque) have a greater risk of coronary death or non-fatal MI.58 Moreover, a large-scale clinical CCTA study (3,158 patients) also indicated that the combination of high-risk plaque characteristics, significant stenosis, and plaque progression by serial examination allows for better prognostic values for future ACS events.59
Intravascular Ultrasound
A number of pathologic studies and clinical papers have proposed the idea of precursor lesion to PR that could be located and treated before it causes coronary events. Catheter-based invasive imaging techniques, including intravascular ultrasound (IVUS), can identify some features of ‘vulnerable plaques’ in atherosclerotic lesions in daily practice, although it cannot visualize exact thinness of the fibrous cap and other more specific details (resolution of IVUS >100 µm). In grayscale IVUS images, coronary plaques can be classified as soft, intermediate, calcified, or mixed according to their echo signals. Additionally, assessment of plaque burden (formula: plaque plus media CSA/external elastic membrane CSA), vessel remodeling index (formula: lesion external elastic membrane CSA/reference external elastic membrane CSA), atheroma eccentricity (formula: [maximum plaque + media thickness − minimum plaque + media thickness]/maximum plaque + media thickness), as well as plaque characterization by reflecting echo signal amplitude, frequency, and power (e.g. integrated backscatter IVUS and virtual histology IVUS.) can provide additional information about potential vulnerable plaque, with the idea that once identified we would be able to treat them before an event can occur.62,72,73 Accordingly, larger plaque burden, eccentric pattern, outward remodeling (remodeling index >1.0), spotty calcification, signal attenuation without dense calcium, presence of an echolucent zone, and plaque composition of necrotic core determined by radiofrequency analysis have been reported to be vulnerable plaque characteristics.74 However, in general, the results of clinical studies have not clearly supported the theory of IVUS-defined TCFAs as the lesion causing clinical events. The PROSPECT trial was a prospective study of 697 ACS patients who underwent three-vessel coronary angiography and gray-scale and radiofrequency IVUS.62 From 595 identified IVUS-defined TCFAs, only 26 sites had a coronary event at a median follow-up of 3.4 years. When combined with other criteria, such as a plaque burden of >70% and a minimum lumen area of <4 mm2, the HR increased to 11.05 for any lesion having these criteria, yet 88.2% of patients harboring these plaques did not go on to have an event during the study. Another clinical study using virtual histology IVUS also revealed similar results, although the number of patients and follow-up duration were lower.63
Optical Coherence Tomography
Optical coherence tomography (OCT) is another major coronary artery invasive imaging modality using near-infrared light and has 10 times greater resolution compared with IVUS (resolution >10 µm). Therefore, fibrous cap thickness can be measured by OCT, and this technology has emerged as the best discriminator of plaque type in autopsy studies.75,76,77 To verify the findings of pathologic autopsy study regarding fibrous cap thickness and other features in coronary plaques, an OCT- and IVUS-based in-vivo clinical study was conducted.23,78 The specific morphological characteristics of ruptured culprit plaque responsible for acute events, ruptured non-culprit plaques (without lumen occlusion), and non-ruptured TCFA were compared in patients with ACS.78 In total 126 plaques from 82 cases were analyzed, and fibrous cap thickness was thinner in ruptured plaques (both culprit [43 ± 11 μm] and non-culprit [41 ± 10 μm] lesions) than in TCFA (56 ± 9 μm, p<0.001 and p<0.001, respectively).
Another OCT-based clinical study involving 643 plaques from 255 patients showed that TCFA were highly prevalent in various stages of coronary atherosclerotic disease suggesting their dynamic nature.79 Compared to mildly stenotic TCFAs, severely stenotic TCFAs showed greater plaque burden as well as more vulnerable plaque features. This suggests severely stenotic TCFAs may be more likely to lead to rupture and thrombosis in the near future. Recently, Kubo et al. demonstrated a prospective OCT study, including 3,533 non-culprit plaques from 1,378 patients.66 Seventy-two ACS arose from initial non-culprit plaques within 6 years of follow-up. A larger maximum lipid arc, thinner minimum fibrous cap thickness, and smaller minimum lumen area were independent risks for subsequent ACS at the lesion level.66 On the other hand, regarding other vulnerable plaque characteristics, such as IPH and microcalcification in the fibrous cap, the performance of OCT remains uncertain (i.e. the former is because of the complex findings during healing of hemorrhage in the necrotic core, and the latter is because of the lower end of the spectrum [microcalcification <5 µm diameter]). In addition, discriminating calcified areas from lipid core in OCT is difficult as both appear as signal-poor regions.80
Near-infrared Spectroscopy
Near-infrared spectroscopy (NIRS) is another clinically available intra-coronary imaging modality using characteristic emission spectra produced by plaque contents following interaction with photons. NIRS can be used to identify lipid core plaque (LCP) as validated in several pre-clinical and clinical studies.81–83 The calculated data by intra-coronary NIRS system provides a two-dimensional map of the vessel (chemogram), i.e. the x- and y-axis represent pullback position and circumferential position in degrees (0–360°), respectively, with a color scale from red to yellow indicating probability for the LCP presence. From the chemogram, a summary metric of the probability of LCP presence in at a 2-mm interval during pullback can be computed and displayed in a color map (block chemogram).84 Lipid core burden index (LCBI) is a quantitative summary metric of the LCP presence in the entire scanned segment, which is the fraction of valid pixel in the chemogram that exceeds an LCP probability of 0.6, multiplied by 1,000. For instance, maxLCBI4mm indicates the maximum value of the LCBI for any of the 4 mm segments in the interrogated region.84 This method is able to quantify the lipid content of plaque, especially in cases of positive remodeling with large lipid-rich necrotic core.85
A recent large-scale clinical trial using NIRS, the LPR study, revealed the usefulness of NIRS-based vulnerable plaque and patient risk stratification.69 A total of 1,563 patients with CAD underwent NIRS-IVUS assessment in two or more non-culprit arteries, and patient- and plaque-level events were enrolled. To examine the association between maximum 4 mm LCBI (maxLCBI4mm) and non-culprit major adverse cardiovascular events, patients with large versus small lipid-rich plaque (maxLCBI4mm≥250 versus maxLCBI4mm<250, respectively) were followed for up for 2 years. The adjusted patient-level analysis found an 18% higher risk of experiencing a non-culprit event within 2 years for each 100 unit increase in LCBI 4 mm segment. More recently the prospective multicenter trial PROSPECT II also examined the usefulness of combined coronary plaque assessment with NIRS + IVUS in 3,629 non-culprit lesions of 898 patients with recent MI.68 Adverse events within 4 years occurred in 112 patients (13.2%), with 66 events (8.0%) that originated from non-culprit lesions. Highly lipid-rich lesions assessed by maxLCBI4mm, large plaque burden, as well as minimum lumen area were independent predictors of non-culprit lesion related MACEs. Although these data suggest plaque assessment by NIRS + IVUS can predict overall risk of events, analysis on an individual plaque basis was not presented. Therefore, on a patient- and plaque-specific basis, the technology could not discriminate the future risk of events in the near term for a specific plaque.
What is the Significance of Vulnerable Plaque Detection by Imaging Modalities?
The fact that even PR events do not always cause acute MI or sudden coronary death and can happen without clinical symptoms complicates the quest to identify vulnerable plaque by imaging modalities in order to prevent clinical events.86,87 Arbab-Zadeh et al. reviewed 11 clinical and pathological studies involving 1,371 patients and identified subclinical PR was detected in the non-culprit lesion in 11.5% of patients with stable CAD or healthy controls, as well as 21.5% of patients who presented with ACS.87
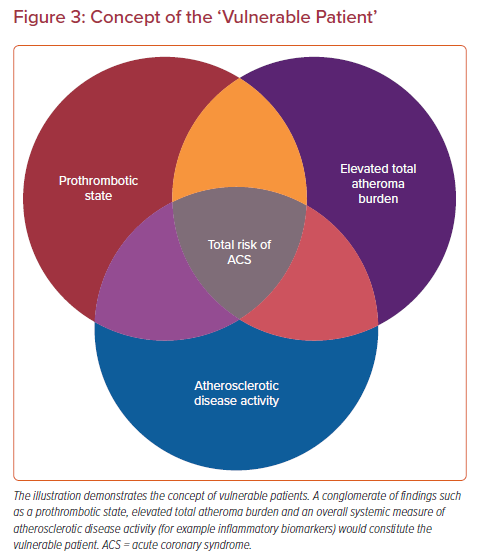
Prior pathological evidence by Mann et al. advanced the theory that repeated silent PRs are one of the critical triggers of phasic rather than linear progression of luminal narrowing in diseased coronary arteries which may involve subclinical thrombus formation and healing.88 Also, a histopathology study from our group involving 142 cases who died of sudden coronary death revealed frequent observation of multiple healed PR sites with layering in the lesion with acute and healed PR.35 Moreover, in patients dying without acute thrombus, healed PR sites were identified in 80% of cases, and cumulative healed PRs at the same location clearly related to increased percentage stenosis. Combined with the evidence from IVUS-based studies that 75% of TCFAs convert into thick-capped fibroatheromas within a 12 month follow-up period, silent PR and healing are not uncommon in vivo.89 Together, this evidence suggests that even if the location of vulnerable plaque was detected precisely, many of them would not cause clinically important symptomatic events. Furthermore, multiple sub-clinical ruptured plaques as well as non-ruptured TCFA have been detected in addition to culprit lesions in patients with ACS, suggesting a systemic condition leading plaque vulnerability throughout the coronary tree.90 The concept of the ‘vulnerable patient’ as a contributor to near-term events in subjects harboring multiple vulnerable plaques needs more attention.91,92
Factors Associated With Acute Coronary Event: Plaque Development and Coagulation Status
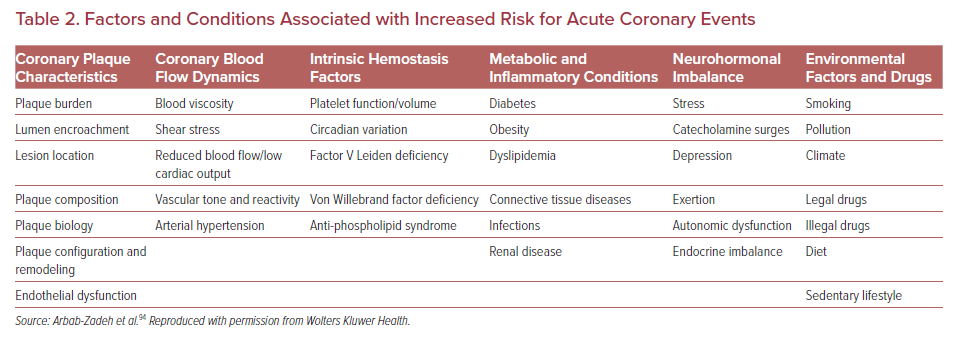
The vast majority of sites identified by imaging as vulnerable plaque (TCFA) do not cause symptomatic arterial thrombotic occlusion. The above-mentioned PROSPECT trial showed that only 4.4% (26/595) of non-culprit TCFA lesions detected by Virtual Histology-IVUS (VH-IVUS) in 313 patients turned in clinically detectable thrombotic events during 3 years follow up.62 Indeed, large number of plaque rupture or erosion event can happen silently (without leading symptomatic coronary events) as shown in pathology and clinical studies.2,93 These silent thrombotic events mainly result in plaque volume progression by intramural thrombus organization rather than life threatening acute coronary occlusion and MI. Therefore, although the release of pro-thrombotic necrotic core components (e.g. tissue factor) to blood stream after fibrous cap disruption in PR or the exposure of pro-thrombotic subendothelial tissue matrix caused by endothelial disruption in PE can be an initial trigger of thrombotic formation. Multiple other factors may also contribute to subsequent thrombus enlargement and final luminal occlusion. Factors and conditions associated with increased acute coronary event risk may be further categorized into being related to plaque characteristics, coronary flow dynamics, intrinsic hemostatic/fibrinolytic dysfunction, neurohormonal dysregulation, and environmental factors and triggers as shown by Arbab-Zadeh et al. (Table 2).94 Therefore, even though the detection of vulnerable plaque is a quite important, for comprehensive understanding of acute coronary event (to recognize ‘vulnerable patient’), identification of factors associated with plaque development/progression, with local/systemic blood procoagulant status, or with combination of them need to be considered. In this regard, cardiovascular physicians need to appreciate these nuances – many of which remain incompletely understood.
Management and Treatment of Coronary Vulnerable Plaque
Lipid-lowering Treatment
A number of clinical trials revealed that the reduction of LDL cholesterol with statins is associated with improvements in fatal cardiovascular events.95 According to the recently available clinical guidelines, lipid-lowering using statins is a standard treatment for the primary and secondary prevention of cardiovascular disease caused by atherosclerosis.96,97 Furthermore, statin therapy is recommended in both ACS and prior to percutaneous coronary intervention (PCI), and this has originated from its pleiotropic effect on several cellular pathways regarding anti-inflammation and subsequent anti-thrombosis.98 A regression of coronary plaque burden by statin therapy was confirmed by observation with IVUS in a pooled analysis of eight clinical trials including 4,477 patients with high-risk plaques.99 The long-term effect of statin on coronary plaque was also confirmed in recent PARADIGM study.100 In this prospective, multinational trial over 2 years of follow-up, statin treatment was associated with slower progression of overall coronary atherosclerosis, a reduction of high-risk plaque characteristics, and increased plaque calcification.100 A reduction of plaque burden and an increase in fibrous cap thickness of coronary fibroatheroma was also confirmed by several OCT-based clinical studies.101 When comparing low- and high-dose statin treatment, high-dose treatment was associated with fewer vulnerable plaque features including lipid volume and fibrous cap thickness confirmed by serial OCT observation.102
In addition to statins, proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors have recently emerged as another intensive lipid-lowering agent.103 Large scale randomized controlled trials confirmed the significant effect of PCSK9 inhibitors on LDL-cholesterol levels as well as cardiovascular event reductions in patients with a background of statin therapy.104,105 The updated European lipid-lowering guideline recommends adding a PCSK9 inhibitor early after the ACS event in patients whose LDL-cholesterol levels have not reached goal despite maximum tolerated statin and ezetimibe treatment.97 The US guidelines consider patients with ACS within 12 months as a very high-risk population. In patients in this category whose LDL-C level is >1.8 mmol/l on maximally tolerated statin and ezetimibe, adding a PCSK9 inhibitor in this setting is reasonable.106 A study of the GLAGOV trial, which investigated 968 statin-treated CAD patients who underwent serial IVUS imaging before and after (i.e. 76 weeks) adding placebo or evolocumab treatment, revealed greater reduction of atheroma volume in evolocumab group.107 However, serial plaque compositions assessed by virtual histology IVUS were similar between evolocumab and placebo groups.107 An ongoing multicenter, double-blind, placebo-controlled trial, PACMAN-AMI, will determine the effect of alirocumab on top of high-intensity statin therapy on high-risk coronary plaque characteristics by serial assessment with intra-coronary multimodalities including IVUS, NIRS, and OCT.108
Anti-inflammatory Treatment
More than two decades ago, the role of inflammation in atherosclerosis was raised as a potentially important target.109 Ridker et al. demonstrated apparently healthy individuals with elevated levels of high-sensitive C-reactive protein (hsCRP) to be at high vascular risk (MI and stroke) irrespective of lipid levels.109 Recently, the inflammatory character of atherosclerosis has come into the forefront with the results of several clinical trials.
The effect of canakinumab, a therapeutic monoclonal antibody targeting interleukin (IL)-1β was tested in a large scale randomized, double-blind trial, CANTOS.110 A total of 10,061 patients with previous MI history with elevated hsCRP were randomly allocated to canakinumab and placebo and followed-up for 3.7 years. The anti-inflammatory therapy achieved a 15% relative reduction in risk for recurrent MI, stroke, or cardiac death. Individuals who responded to canakinumab therapy by accomplishing a greater than median reduction in hsCRP had a 26% reduction in the primary end point and a decrease in all-cause mortality. Because IL-1β participates in host defenses, canakinumab treatment was also associated with a higher incidence of fatal infection than placebo. Thus, the data did not completely support the use of anti-inflammatory medicines as a method to decrease plaque vulnerability, and the Food and Drug Administration did not support labeling for canakinumab as a targeted therapy for reduction of cardiovascular events.
Another anti-inflammatory agent, methotrexate, has also been tested for efficacy in cardiovascular risk prevention in a large scale, randomized, double-blind trial, and the result was recently reported.111 However, low-dose methotrexate did not reduce levels of IL-1β, IL-6, or CRP and did not result in fewer cardiovascular events than the placebo.
Colchicine is an alkaloid derivative that has been traditionally used for treating gout as well as several other rheumatic diseases.112 Growing evidence suggests that colchicine’s anti-inflammatory mechanisms, such as NLRP2 inflammasome inhibition, can be a treatment option for atherosclerotic disease. The effect of colchicine treatment in patients with stable CAD on top of statin and antiplatelet therapy was tested in two randomized clinical trials: LoDoCo and LoDoCo-2.113,114 The latter trial had a much larger population of 5,522 patients with stable CAD on statin and antiplatelet therapy who were allocated to colchicine versus placebo. Those who received colchicine had a 31% risk reduction in primary outcomes, including cardiovascular mortality, myocardial ischemia, ischemia-driven revascularization, and stroke compared with the placebo (p<0.05).114 As opposed to canakinumab in the CANTOS trial, colchicine used in this study was not associated with an increased rate of hospitalization for infection. Moreover, colchicine’s effect on secondary cardiovascular prevention was evaluated in the large scale COLCOT trial.115 In this study, 4,745 patients who had suffered a recent MI were randomly assigned to two groups: low dose colchicine or placebo. The colchicine group had a significant risk reduction (23%) in the primary composite end point, i.e. cardiovascular death, cardiac arrest, MI, stroke, or urgent coronary revascularization. Considering the inflammatory characteristics of rupture-prone vulnerable plaques, anti-inflammatory treatment might decrease the risk of rupture, but their routine use in this setting likely requires more data.
Invasive Treatment
As shown in the ISCHEMIA trial, our current understanding of the effectiveness of a PCI strategy added to guideline-directed medical therapy in patients with stable CAD is questionable, with likely no beneficial effect on cardiovascular outcomes compared to medical therapy alone.116 On the other hand, a recently reported meta-analysis analyzed 46 trials, including 37,757 patients, and revealed a beneficial effect of PCI in unstable patient scenarios including: post-MI patients who did not receive immediate revascularization; patients who underwent primary PCI for ST-elevation MI but with residual coronary lesions; and patients with non-ST-elevation MI.117 Overall, in these unstable scenarios, PCI reduced all cause of death (RR 0.84; 95% CI [0.75–0.93]; p=0.02), cardiac death (RR 0.69; 95% CI [0.53–0.90]; p=0.007) and MI (RR 0.74; 95% CI [0.62–0.90]; p=0.002). Patients undergoing coronary angiography often present with multi-vessel disease, and the finding of vulnerable plaques is common under observation with in-vivo imaging modalities, especially in unstable scenarios. However, to date, there is no evidence that prophylactic revascularization of vulnerable plaques is beneficial, and the significance of imaging techniques in the assessment of vulnerable plaques still needs more evidence to be routinely considered as necessary. The CANARY trial tested the correlation between lipid rich plaque as detected by NIRS and periprocedural MI presumably because of distal embolization.118 Eighty-five patients were enrolled at nine US sites. NIRS-IVUS were performed at baseline, and lesions with a maxLCBI4mm >600 were randomly allocated to PCI with or without distal protection filter. Periprocedural MI developed in 21 patients (24.7%) and the maxLCBI4mm was significantly higher in patients with versus without MI (p<0.05). Although the beneficial effect of distal protection was not confirmed in 31 randomized lesions with maxLCBI4mm >600 probably because of the lack of study power, the attempt to perform tailor-made PCI treatment with plaque characterization by contemporary coronary imaging needs further evaluation.
As mentioned above, current OCT imaging allows us to identify underlying plaque morphology in ACS. Compared with PR, the culprit lesion of PE typically has larger lumen, preserved vascular structure, and platelet-rich thrombus.13,15,119 Because drug-eluting stent implantation has the intrinsic disadvantages (e.g. the development of neoatherosclerosis, limiting normal vasomotion, and preclusion of bypass surgery due to metallic caging120,121) conservative management with antithrombotic therapy (particularly antiplatelet therapy) without stenting can be a management option for ACS caused by PE. Several small clinical trials have shown the feasibility of this treatment strategy in short- and long-term outcomes.122,123 Large-scale randomized trials are needed to replicate these pilot study results and to further confirm long-term outcomes of this new treatment option in patients with ACS caused by PE.
Conclusion
The vulnerable plaque concept originating from human pathology studies has led to a remarkable advancement in our understanding of pathogenesis and treatment for atherosclerosis. We know, to date, that comprehensive risk assessment needs to be performed to identify patients at risk for adverse cardiovascular events (Figure 3). Developments of invasive and non-invasive coronary imaging techniques are allowing for the detailed detection of rupture-prone vulnerable plaque characteristics. In combination with cardiovascular risk factors, plaque imaging might provide a more accurate picture of an individual’s risk.
The diagnostic value of currently available intra-coronary imaging (e.g. IVUS, OCT, and NIRS) is extraordinary as many interventional cardiologists have already recognized. However, the use of intra-coronary imaging in daily practice is still low, especially in western countries.124 Its high cost, the prolongation of procedure time, and risk of complications are all potential reasons for the low adoption.124 As mentioned, a number of clinical studies have demonstrated the usefulness of current intra-coronary imaging modalities not only to define plaque characterization but also to facilitate the optimal stenting process. Also, regarding economic impact, the favorable incremental cost-effectiveness of IVUS-guided PCI versus angiography-guided PCI has been reported in several studies.125,126 Therefore, more use of intra-coronary imaging in daily clinical PCI procedures should be encouraged to achieve better clinical outcomes. Reimbursement issues as well as complexity in its interpretation (sometime requiring significant expertise) remain as additional barrier to its widespread adoption.
According to the results of large-scale clinical trials, the effectiveness of invasive PCI strategies in stable CAD patients is quite limited. Unstable patient populations who might harbor more vulnerable plaques need to be stratified, and more intensive medical therapeutic approaches need to be considered. As of now, imaging is not able to determine with high accuracy on a patient- or lesion-basis who is at high-risk in the near term for PR. More work needs to be done to understand other features that determine plaque vulnerability beyond those determined by morphology. In addition, the current approach of imaging-based vulnerable plaque detection is mainly focused on rupture prone lesions, which are represented as TCFA in pathology. Since the precursor lesion of PE and CN (more than 30% of the population in the case of ACS) has not been determined yet, further basic and clinical studies, including coronary imaging, need to be advanced for better risk stratification in patients with CAD.