








Decades ago heart failure (HF) was primarily regarded as a hemodynamic disorder in an attempt to explain patients’ symptoms and disability. This hemodynamic model led to the widespread evaluation of peripheral vasodilators (to increase cardiac output by decreasing systemic vascular resistance against the failing heart). It also led to the development of novel positive inotropic agents (to directly increase cardiac output). Long-term use of these drugs failed to improve symptoms and was frequently accompanied by an increase in the risk of death. 1–3 These clinical observations raised concerns about the validity of the hemodynamic hypothesis and led to the development of alternative models of HF—most importantly, the neurohormonal hypothesis.
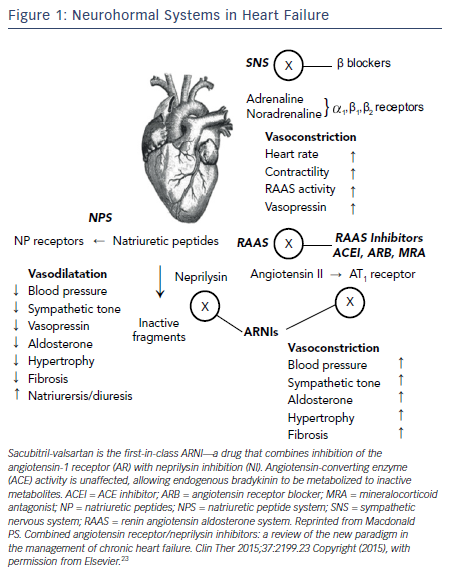
It is now well understood that chronic HF is associated with a complex pattern of neurohormonal activation that contributes to the relentless progression of this fatal syndrome. 4 The theme of these systems is that initially beneficial mechanisms are ultimately maladaptive. The activation of the sympathetic nervous system (SNS) and the renin-angiotensin- aldosterone system (RAAS) initially cause vasoconstriction and fluid retention to maintain perfusion in the face of a falling cardiac output from the failing heart. While effective in the short term, the long-term effects of SNS and RAAS activation are adverse cardiac remodeling leading to progressive cardiac dilatation and further dysfunction of the failing heart (see Figure 1 ).
The current guideline-directed medical therapy for HF is, in essence, a triumph of translation medicine. An understanding of this neurohormonal activation, translated into medications to block these maladaptive systems, has culminated in improved quality of life (QoL) and survival for patients with HF. 5 The most effective drug therapies for chronic systolic HF are those that inhibit the activity of the SNS and RAAS. These agents include the beta-blockers to inhibit SNS activity and the angiotensin-converting enzyme inhibitors (ACEIs), angiotensin II type 1 receptor blockers (ARBs), and mineralocorticoid receptor antagonists (MRA) to act on the RAAS pathway. Recently, however, two new promising therapies for HF have emerged: ivabradine and the neprilysin inhibitors. Each capitalizes on the current paradigm of neurohormonally-focused therapies to improve outcomes in HF patients. This article will review the current management of HF patients and provide insight into the rationale behind these two new promising therapies for HF.
Guideline-directed Medical Therapy for Heart Failure
RAAS Blockers
In the current era it is difficult to imagine a time when ACEIs were not standard of care for HF patients. However, prior to the publication of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS) in 1987, this was not the case. In the landmark CONSENSUS trial,6 patients with New York Heart Association (NYHA) Class IV symptoms were randomized to enalapril or placebo. The trial was terminated early after 253 patients had been randomized and an average follow-up of 188 days. Six-month mortality in the enalapril group was 26 % compared with 44 % in the placebo group, giving a relative risk-reduction of 40 % (P=0.002). At 1 year these proportions were 36 and 52 % (P=0.001). While the trial is decades old, these figures are important to consider; the placebo group mortality highlights the dreadful prognosis in patients with severely symptomatic HF before the advent of modern disease-modifying therapies.
In multiple trials to follow, the benefit of ACEIs has been demonstrated in patients with less symptomatic HF,7,8 asymptomatic HF,9 and HF after an acute myocardial infarction.10 The trials of ARBs have been less definitive. While ARBs are clearly better than placebo,11 they do not appear superior to ACEIs for patients with HF.12 The combination of ACEI and ARB results in adverse effects of hypotension and hyperkalemia.13 However, the MRAs spironolactone and eplerenone have shown great promise in HF patients. The rationale behind the use of MRAs is that although ACEIs reduce the secretion of aldosterone, an “escape” phenomenon may occur and, in addition, aldosterone secretion is also independently controlled by serum potassium concentration and corticotrophin. The benefits of MRAs in addition to ACEIs have been shown in patients with severe symptomatic H F,14 less symptomatic HF,15 and HF after acute myocardial infarction.16
Beta-blockers
The role of beta-blockers in the management of HF is an even more extraordinary example of the power of translation medicine. While one could understand the benefit of ACEIs or ARBs on a hemodynamic as well as neurohormonal level, accepting the benefit of a negative inotrope in the survival of HF patients truly required an acceptance of the maladaptive consequences of SNS activation. The most striking of all the beta-blocker trials in HF is the Carvedilol Prospective Randomized Cumulative Survival trial (COPERNICUS), which randomized patients with ambulatory NYHA Class IV symptoms and ejection fraction (EF) < 25 % to carvedilol or placebo, on the background of treatment with ACEI. In 2,289 patients followed for less than a year, there was a 35% reduction in mortality. Thus, not only did the most advanced HF patients tolerate beta-blockade in this trial, they also derived survival benefit from it. The benefit of beta-blockers has also been shown in less severe HF17,18 and HF after acute myocardial infarction.19
Ivabradine
Effect of Heart Rate on Survival
While beta-blockers are clearly beneficial in HF patients, there is evidence that the degree of heart rate lowering affects survival. In a subgroup analysis of the Morbidity-mortality Evaluation of the lf inhibitor ivabradine in patients with coronary disease and left ventricular dysfunction (BEAUTIFUL) trial comprising patients who were aged 55 and older with coronary artery disease and EF <40 % there was a strong correlation between elevated baseline resting heart rate and mortality. A baseline resting heart rate of <70 beats per minute (bpm) was associated with an increased 34 % increased risk of cardiovascular death and a 53 % increased risk of HF admission. Furthermore, for every increase of 5 bpm in baseline resting heart rate, there was an 8 % increase in cardiovascular death and a 16 % increase in admission to hospital for HF (P<0.0001).20 Similar results were demonstrated in a meta-analysis of beta-blocker HF trials including almost 23,000 patients.21
Of course, an association between lower heart rate in HF patients taking an evidence-based beta-blocker and survival does not imply causation: does a lower heart rate result in improved survival, or is a lower heart rate simply a marker of better prognosis? In addition, up-titration of beta-blockers in HF patients may be limited by hypotension and fatigue from the acute negative inotropic effects. However, a medication that could lower heart rate without affecting contractility would be the perfect test to this hypothesis. Ivabradine, through the Systolic Heart failure treatment with the lf inhibitor ivabradine Trial (SHIFT) study, has provided an opportunity to do so.
The SHIFT Study
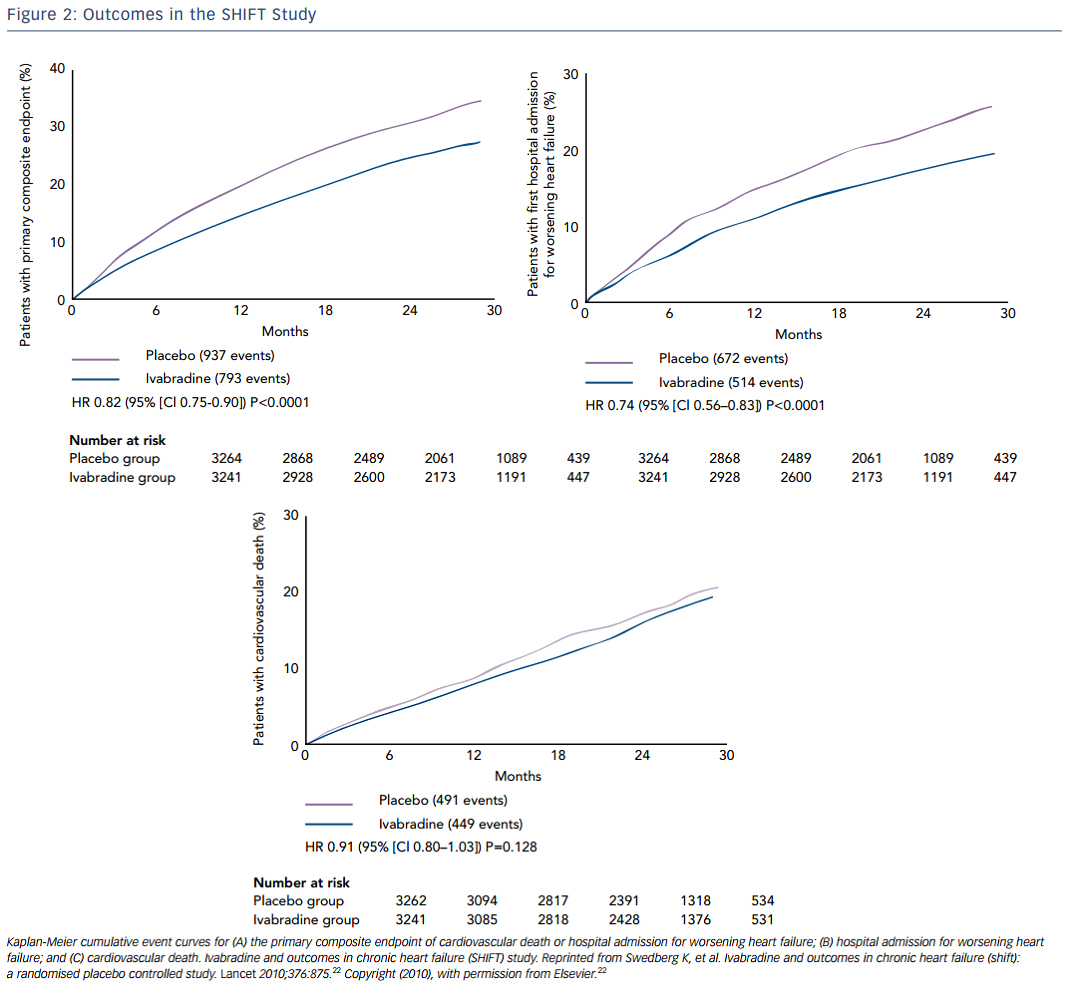
Ivabradine is a selective inhibitor of the hyperpolarization-activated cyclic-nucleotide-gated funny current (involved in pacemaker-generation and responsiveness of the sinoatrial node, which results in heart-rate reduction with no other apparent direct cardiovascular effects. Thus, ivabradine decreases heart rate without decreasing contractility. In the SHIFT study, 6,558 patients with EF ≤35 %, heart rate ≥70 bpm, at least one HF hospitalization, and taking maximally-tolerated beta-blockers were randomly assigned to ivabradine or placebo. Over almost a 2-year follow-up, ivabradine resulted in a significant reduction in the primary endpoint of cardiovascular death or hospital admission for worsening HF, which was mainly driven by reductions in hospitalizations for HF. HF hospitalization was 21 % in the placebo group and 16 % in the ivabradine group (see Figure 2 ).22
Prescribing Considerations for Ivabradine
In April 2015 the US Food and Drug Administration (FDA) granted approval of ivabradine (Corlanor ® ). It was approved for use to reduce the risk of hospitalization for worsening HF in patients with stable, symptomatic chronic HF with EF ≤35 %, who are in sinus rhythm with a resting heart rate of ≥70 bpm, and are either on maximally tolerated doses of beta-blockers or have a contraindication to beta-blocker use.
Ivabradine is given as a starting dose of 5 mg twice daily and can be increased to 7.5 mg twice daily after 2 weeks in patients whose heart rate remains over 60 bpm. The most important consideration in deciding to prescribe ivabradine is that it is not a substitute for beta-blocker therapy. Beta-blockers have clear survival benefit in HF whereas ivabradine, in conjunction with maximally tolerated beta-blocker dosing, mainly reduces HF hospitalizations. Ivabradine is contraindicated in patients with acute decompensated HF, blood pressure <90/50 mmHg, resting heart rate below 60 bpm prior to treatment, or pacemaker dependence. The most common adverse reactions include bradycardia, hypertension, atrial fibrillation, and luminous visual phenomena (phosphenes).
Neprilysin Inhibitors
The Natriuretic Peptide System and other Endogenous Vasoactive Peptides
While activation of the SNS and RAAS are maladaptive in HF, activation of the natriuretic peptide system (NPS) is beneficial (see Figure 1 ). The NPS includes a family of three peptides known as atrial NP (ANP), brain NP (BNP; initially identified in brain but mainly secreted from ventricular myocardium), and C-type NP. NPs are released into the circulation from the atria (ANP) and from the ventricles (BNP), respectively, in response to atrial and ventricular distension caused by myocardial injury or overload. ANP and BNP exert a range of activities that could be beneficial in HF including vasodilatation, natriuresis, inhibition of the RAAS and SNS, and activation of the parasympathetic nervous system.23 In healthy subjects, ANP and BNP are rapidly cleared from the circulation via either binding to clearance receptors or degradation by an enzyme called neutral endopeptidase, also known as neprilysin.
There has been longstanding interest in the development of drug therapies that either mimic or prolong the activity of endogenous NPs. One approach has been to develop synthetic analogues of ANP and BNP, carperitide and nesiritide, respectively. Both drugs require intravenous administration and have a short duration of action, necessitating continuous infusion rather than bolus administration. Continuous infusion has limited the clinical application of these agents to the treatment of acute decompensated HF and no long-term benefit has been shown.24
A second approach to augmenting the activity of the NPS has been to inhibit neprilysin, with the aim of prolonging the activity of the endogenous NPs that are already elevated in patients with HF. Neprilysin, a neutral endopeptidase, degrades several endogenous vasoactive peptides including natriuretic peptides, bradykinin, and adrenomedullin. Inhibition of neprilysin increases the levels of these substances, countering the neurohormonal over-activation that contributes to vasoconstriction, sodium retention, and maladaptive remodelling. The relative role of natriuretic peptides versus the other agents is not clear but it is uncertain that augmentation in NPs is the sole cause for the benefits of neprilysin inhibition. Interestingly, a neprilysin inhibitor alone is ineffective because it blocks not only degradation of NP and other potentially beneficial vasoactive peptides but it also degrades adrenomedullin, substance P, angiotensin I and II, bradykinin, and endothelin-1. Thus, although inhibition of neprilysin would increase levels of NPs and other compensatory peptides, leading to the desirable effects noted above, there may also be increased levels of peptides, yielding undesirable effects such as vasoconstriction with angiotensin II and endothelin-1.
If a neprilysin inhibitor should be combined with a RAAS blocker, an ACEI would be an obvious choice since ACEI are the cornerstone of RAAS blockade and HF management. However, the combined inhibition of ACEI and neprilysin was associated with serious angioedema in clinical trials.25 This is not surprising since ACEI block degradation of bradykinin and neprilysin inhibitors too. An increase in bradykinin, an inflammatory peptide, could result in angioedema and thus administration of ACEI and a neprilysin inhibitor is contraindicated. The next best approach to RAAS blockade is an ARB, which has set the stage for a new class of drugs: the angiotension receptor-neprilysin inhibitor (ARNI) in HF (see Figure 1 ).

PARADIGM-HF Study
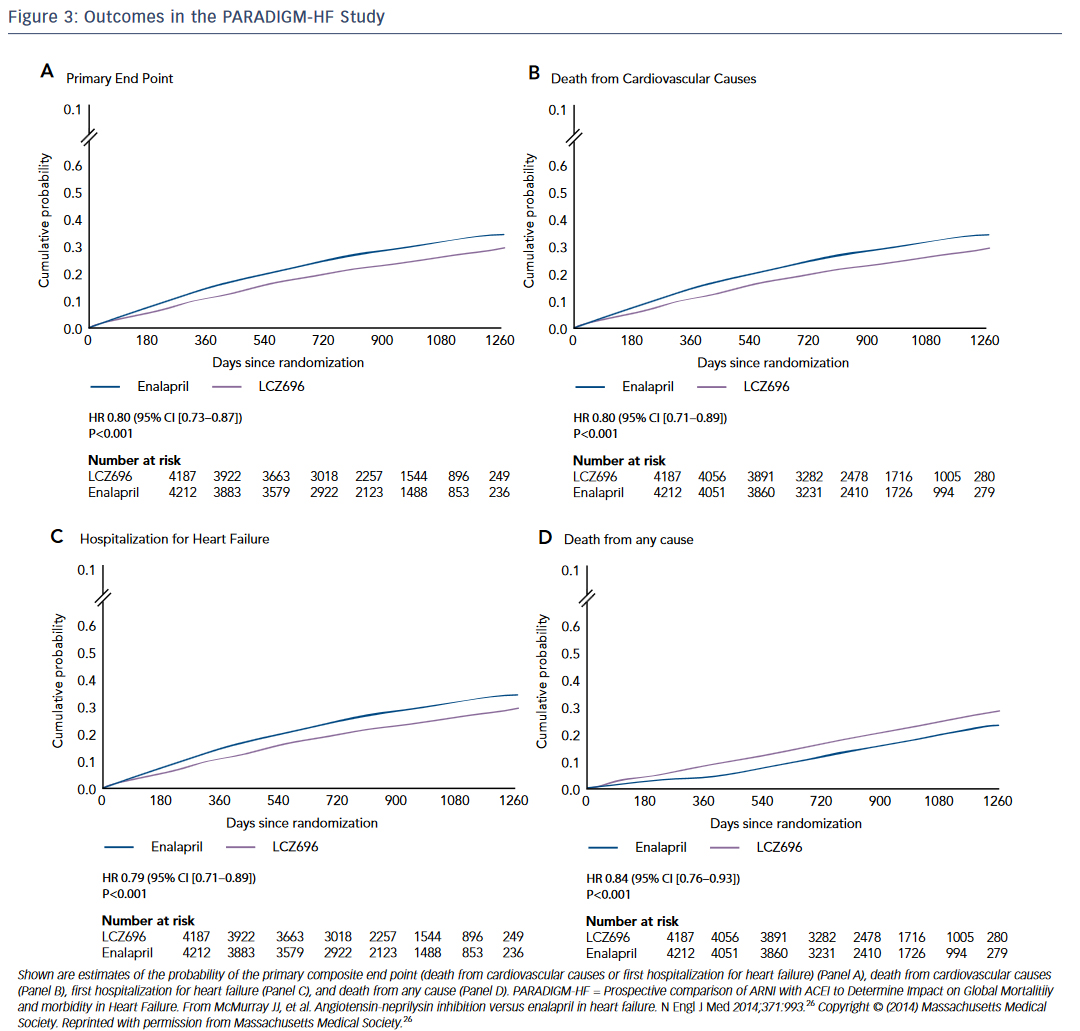
The Prospective comparison of ARNI with ACEI to Determine Impact on Global Mortalitiiy and morbidity in Heart Failure (PARADIGM-HF) study was a randomized trial of the neprilysin inhibitor sacubitril and the angiotensin receptor blocker (ARB) valsartan compared to enalapril in HF.26 The trial was stopped early at 27 months due to overwhelming evidence of benefit in the sacubitril/valsartan arm. Patients receiving sacubitril-valsartan demonstrated a reduction in death and HF hospitalizations (see Figure 3 ). This finding is especially remarkable because sacubitril/valsartan was compared to an active control, not placebo. This is underscored by the speed with which the FDA approved this ANRI. The study was published in August 2014 and by July 2015 the first-in-class drug combination was approved for use in chronic HF under the trade name Entresto.
Prescribing Considerations for Angiotensin-neprilysin Inhibitors
Sacubitril/valsartan is FDA approved to reduce the risk of cardiovascular death and hospitalization for HF in patients with chronic HF (NYHA Class II-IV) and reduced ejection fraction. The recommended starting dose is 49/51 mg (sacubitril/valsartan) twice daily. The dose can be increased after 2–4 weeks to the target maintenance dose of 97/103 mg (sacubitril/valsartan) twice daily, as tolerated. A reduced starting dose of 24/26 mg (sacubitril/valsartan) twice daily should be used for: 1) Patients not currently taking an angiotensin-converting enzyme inhibitor (ACEi) or an angiotensin II receptor blocker (ARB) or previously taking a low dose of these agents; 2) patients with severe renal impairment; or 3) patients with moderate hepatic impairment. The mg dosages of the two drugs are intentionally distinct because two-drug combinations cannot have the same mg amount, as dictated by the FDA.
Sacubitril/valsartan is contraindicated in patients taking an ACEI and must be off an ACEI for at least 36 hours prior to initiation of sacubitril/ valsartan. In addition, sacubitril/valsartan should not be administered to patients with a prior history of angioedema to an ACEI or ARB. The most common adverse reactions are hypotension (expected since both blocking the RAAS and potentiating the NPS causes vasodilation) and hyperkalemia due to the ARB component.
Conclusion
Guideline-directed medical therapy for HF is a triumph of translation medicine whereby an understanding of neurohormonal mechanisms of the RAAS and SNS has resulted in medications that improve QoL and survival in HF patients. Now, increased understanding of the SNS and breakthroughs in capitalizing on the NPS and other compensatory peptides has resulted in two additions to the HF armamentarium, ivabradine and sacubitril/valsartan. One can anticipate that future HF guidelines will incorporate these two new agents into the treatment algorithm and one proposed approach is shown in Figure 4 . Moving forward, the goal of physicians who treat HF patients should be to ensure that all eligible patients receive these exciting new therapies.