







Myocarditis is an inflammatory disease of the myocardium. It has a wide spectrum of severity, with symptoms ranging from mild fevers, shortness of breath, and palpitations to severe hemodynamic collapse. Fulminant myocarditis is a rare form of severe myocarditis that presents with sudden-onset acute heart failure, cardiogenic shock, or life-threatening arrhythmias.1 Prognosis and management of this type of myocarditis varies based on endomyocardial biopsy histopathology, and is classified according to diagnostic criteria initially developed in 1986.2 The microscopic appearance of the myocardial inflammation forms the basis for this classification system.3 The most common microscopic type, and the one with the greatest likelihood of recovery, is lymphocytic myocarditis. More rare, and often more fatal, forms are giant cell myocarditis and eosinophilic myocarditis. In this article, case examples of these three forms of adult fulminant myocarditis will be presented, as well as a review of the current literature on their causes and therapy recommendations.
Lymphocyte Myocarditis
A 61-year-old woman with a history of Crohn’s disease presented with several days of palpitations and shortness of breath. ECG was noted for sinus tachycardia, low voltage, and frequent non-sustained ventricular tachycardia. Troponin T was elevated to 0.09 ng/ml. An echocardiogram showed severe biventricular failure and a left ventricular ejection fraction (LVEF) of 23 %. Angiography was noted for a 70 % mid-left anterior descending artery lesion. Right heart catheterization revealed cardiogenic shock and elevated filling pressures out of proportion for ischemic heart disease.
The patient was initiated on inotropes for supportive care but continued to clinically deteriorate, with subsequent cardiac arrest from ventricular tachycardia. An endomyocardial biopsy was then obtained, which revealed fulminant lymphocytic myocarditis, and she was placed on extracorporeal membrane oxygenation (ECMO) support in the veno-arterial configuration. Further investigation did not identify any precipitant. She was treated with a slow steroid taper and several doses of intravenous gamma-globulin (IVIG) and plasmapheresis. Mechanical support was discontinued on day 30 after she experienced partial recovery. Unfortunately, she eventually expired from complications of her disease.
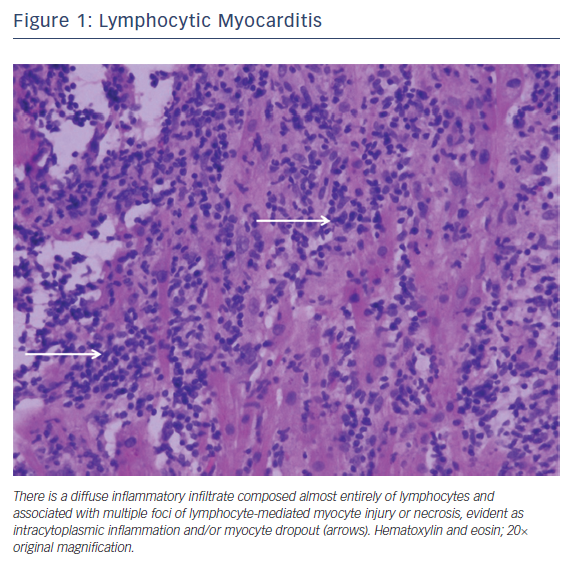
Lymphocytic myocarditis is the most common form of fulminant myocarditis. It is often triggered by a viral infection, either as a primary or secondary immune response.4 Of the viruses, coxsackie B virus and the cytomegaloviruses are the most frequently reported.5 Other infectious causes include bacterial, fungal, and parasitic infections. Medications, toxins, and autoimmune diseases (Takayasu’s disease and granulomatosis with polyangiitis) have also been described. Endomyocardial biopsy can be considered if there is a failure to respond to supportive care or if there is concern over an alternative diagnosis.6 Characteristic histopathology reveals an exclusively or predominately lymphocytic infiltrate with inconspicuous numbers of plasma cells, macrophages, and/or neutrophils (Figure 1).1
As in the case above, the most widely employed therapy for lymphocytic myocarditis is glucocorticoid therapy. However, while a systematic Cochrane review found that steroid therapy may improve LVEF, the use of glucocorticoids in lymphocytic myocarditis has not been shown to reduce mortality.7 Similarly, the use of antiviral therapy, plasmapheresis or IVIG is not routinely recommended due to insufficient human data in the adult population.8,9 This recommendation differs from the pediatric population, in which there is some evidence for the use of IVIG in the treatment of lymphocytic myocarditis.10 This difference may reflect the wider variety of inflammatory etiologies seen in the pediatric population.
Small adult studies do suggest some clinical benefit from combination immunosuppression therapy, such as azathioprine in combination with steroids. Unfortunately, these studies were underpowered and predominately from the era before mycophenolate mofetil, which has generally supplanted the use of azathioprine since its introduction in 1995. The true effectiveness of any of these treatments, though, is difficult to ascertain because of the high rate of spontaneous recovery with supportive care in these patients.11
Giant Cell Myocarditis
A 70-year-old man without prior cardiac history presented with 3 days of progressive dyspnea, orthopnea, and fatigue. ECG showed a new trifascicular block and a Troponin I elevation of 23.78 ng/ml. An echocardiogram revealed a new global cardiomyopathy with an LVEF of 35–40 %. Shortly after admission, he experienced complete heart block, ventricular tachycardia, and sinus arrest. Urgent angiography showed non-obstructive coronary artery disease and right heart catheterization was consistent with biventricular heart failure and cardiogenic shock. Endomyocardial biopsy demonstrated giant cell myocarditis. The patient was initiated on immunosuppressive therapy with steroids and thymoglobulin, and maintained on cyclosporine and mycophenolate. Once stabilized, he underwent implantable cardiac defibrillator (ICD) for secondary prevention. As an outpatient, he continued on standard heart failure medications, steroid taper, cyclosporine, and mycophenolate therapy.
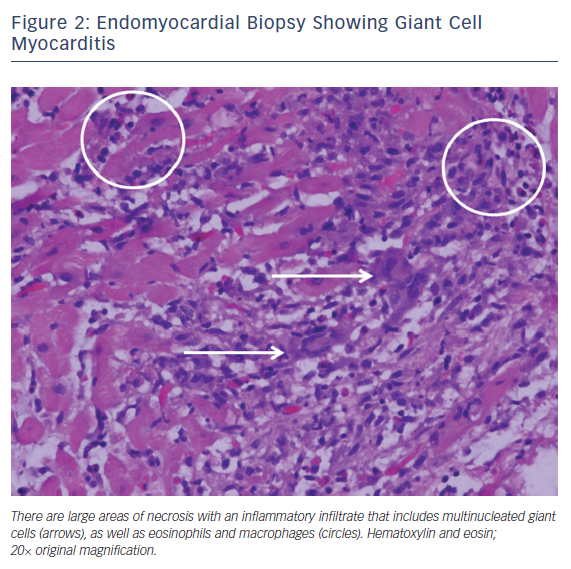
Giant cell myocarditis is a rare, often fatal disease that tends to affect middle-aged adults. It is classically characterized by fulminant heart failure, ventricular arrhythmias and atrioventricular block. The myocardial disease is mediated by T lymphocyte inflammation and it has an association with systemic autoimmune diseases such as thymomas and inflammatory bowel disease. Endomyocardial biopsy is recommended if there is clinical suspicion for giant cell myocarditis.12,13 The characteristic histopathology reveals a mixed inflammatory infiltrate containing lymphocytes, plasma cells, macrophages, and eosinophils, along with numerous multinucleated giant cells without granulomas. This is typically associated with widespread myocyte necrosis, often of the coagulative type (Figure 2).1,13
Treatment of giant cell myocarditis is aimed at attenuating T-cell function. Combination therapies with prednisone, thymoglobulin, cyclosporine, and azathioprine have been used. Alternatives such as mycophenolate mofetil, methotrexate, or the T-cell antibody muromonab-CD3 have also been utilized.1 Discontinuation of prednisone therapy at 6–12 months can be considered if the patient has been medically stable.14 Unfortunately, giant cell myocarditis continues to have a high mortality rate, either from fulminant heart failure or ventricular arrhythmias. In the patients enrolled in the Multicenter Giant Cell Myocarditis Study who were treated with immunosuppressive therapy, the average survival time was 12.3 months.15 Of those who underwent cardiac transplantation, 26 % experienced recurrent giant cell infiltrate of their transplanted heart. Based on the 2017 American Heart Association/American College of Cardiology/ Heart Rhythm Society criteria for appropriate use, in patients with giant cell myocarditis and malignant ventricular arrhythmias, an ICD may be considered (class IIb recommendation) if life expectancy is greater than 1 year.16
Eosinophilic Myocarditis
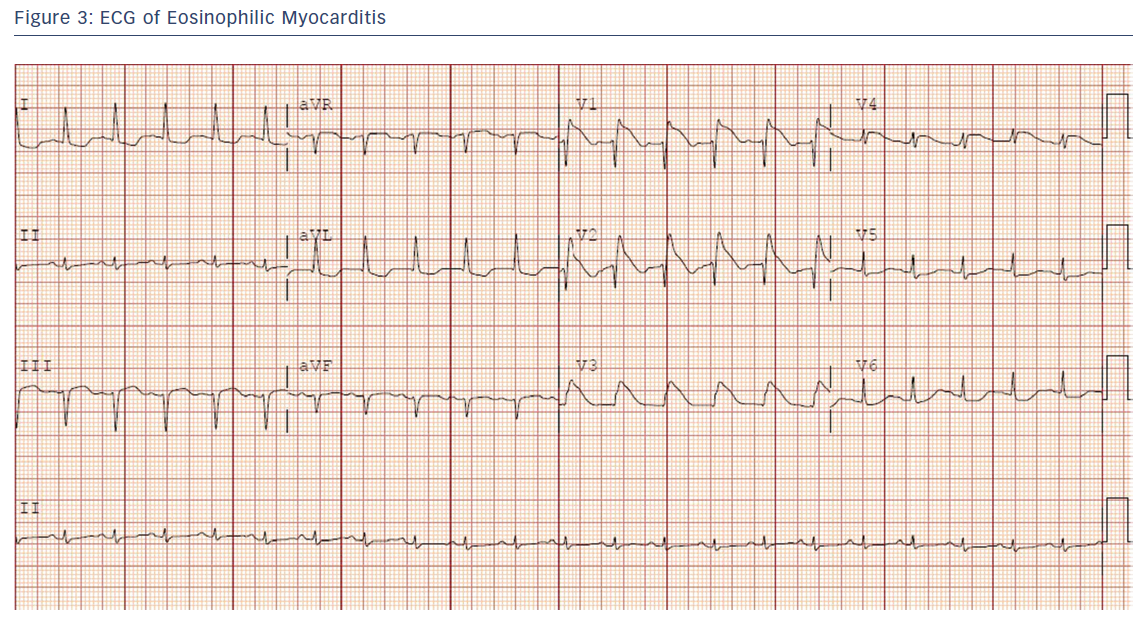
A 30-year-old healthy woman presented with several days of chest pain and fevers. An admission ECG was notable for pronounced anterior ST elevations (Figure 3).
Troponin I was elevated to 10.77 ng/ml and her echocardiogram revealed a globally reduced LVEF of 25 %. She experienced sustained ventricular tachycardia with hemodynamic instability. Angiography revealed normal coronary arteries, and a right heart catheterization was consistent with biventricular failure and cardiogenic shock. She underwent emergent placement of ECMO in the veno-arterial configuration. Peripheral eosinophil count was 0 but endomyocardial biopsy demonstrated diffuse eosinophilic invasion of the myocardium, and a diagnosis of acute eosinophilic myocarditis was made. Further investigation did not identify any precipitant of her myocarditis. She was treated with a slow steroid taper. Mechanical support was able to be discontinued after 6 days, when an echocardiogram showed recovered biventricular function. As an outpatient, she continued on standard heart failure medications and steroid taper without recurrence of her heart failure.
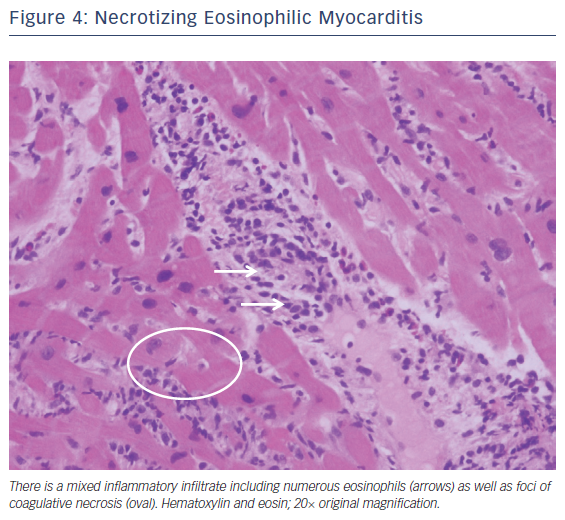
Fulminant eosinophilic myocarditis is a very rare myocarditis mostly reported in adolescents and young adults. It can present as cardiogenic shock, conduction disease, or acute myocardial infarction.17 It can be associated with drug reactions, parasitic infections, hematologic cancers, hypereosinophilic syndrome, or eosinophilic granulomatosis with polyangiitis. Still other cases remain idiopathic or undefined.18 The presence of peripheral eosinophilia should heighten the suspicion for eosinophilic myocarditis, but in one review only 75 % of biopsy-confirmed eosinophilic myocarditis demonstrated any peripheral eosinophilia.18,19 Endomyocardial biopsy can be considered if the clinical diagnosis is in question. The characteristic histopathology of eosinophilic myocarditis is of a mixed inflammatory cell infiltrate containing prominent numbers of eosinophils without giant cells (Figure 4).20
Treatment is focused on timely identification of the disease, treatment of underlying etiologies or withdrawal of any causative agents, and supportive care.19 However, as in the case presented, the etiology is not always apparent.20 Given the underlying inflammatory nature of the disease, treatment with high-dose corticosteroids has had documented success, although no randomized controlled trials have been conducted.21,22 Additional immunosuppressive therapy has been used but remains controversial. While recognition of eosinophilic myocarditis is improving, data regarding its recurrence, risk of future ventricular arrhythmias, and development of post-inflammatory dilated cardiomyopathy are still lacking.18 Therefore, prophylactic ICD placement is not recommended at this time.16
Summary
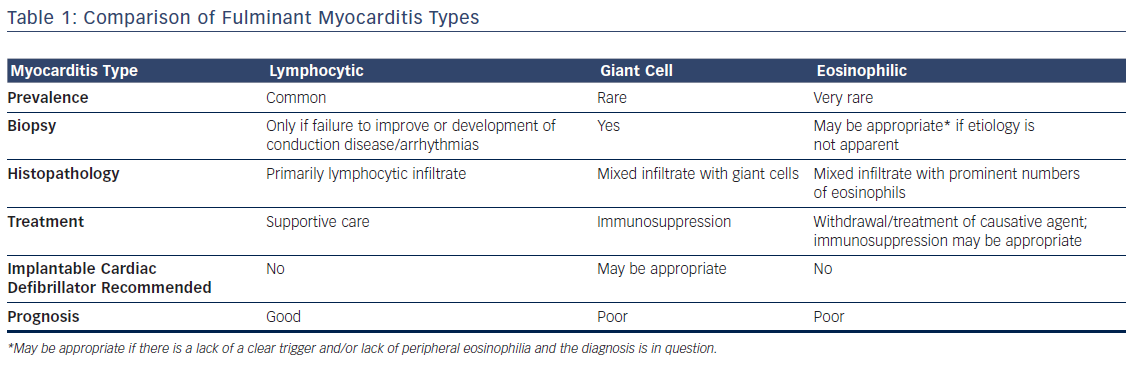
The differences between the three types of fulminant myocarditis are summarized in Table 1.
Survival in fulminant myocarditis depends on timely identification and implementation of advanced circulatory support. Biventricular dysfunction remains the main predictor of death or need for transplantation.21,22 Endomyocardial biopsy is recommended for all patients (class I recommendation) presenting with fulminant heart failure of less than 2 weeks’ duration with hemodynamic compromise, new-onset heart failure of 2 weeks’ to 3 months’ duration with dilated cardiomyopathy, new ventricular arrhythmias, second- or third-degree heart block, or failure to respond to usual support care within 1–2 weeks.6,23. Endomyocardial biopsy can be considered for those with eosinophilia and recent-onset heart failure with potential allergic reaction (class IIa recommendation). Immunosuppression has a potential therapeutic role but requires further investigation before a standardized approach can be recommended. Clinicians should be suspicious for fulminant myocarditis in any patient presenting with new-onset malignant arrhythmias, conduction disease, or sudden cardiovascular collapse.