







After a drought in viable new therapies for more than a decade, two new drugs for use in heart failure patients were approved by the US Food and Drug Administration (FDA) in 2015. These drugs represent two new classes of agents in the heart failure space: a combined angiotensin receptor-neprilysin inhibitor (ARNI) (sacubitril/valsartan; brand name Entresto®, Novartis) and a sinoatrial node modulator (ivabradine; brand name Corlanor®, Amgen). Both drugs are recommended for use as part of a comprehensive medical therapy regimen. The clinical trials for, respectively, Novartis’ Entresto and Amgen’s Corlanor were so strong in their positive results for heart failure patients, that the American College of Cardiology/American Heart Association/Heart Failure Society of America (HFSA) and the European Society of Cardiology produced focused updates in 2016 highlighting the use of both drugs and adding criteria to their respective guideline documents.1,2 ARNI received a class I, level of evidence B-R recommendation, while ivabradine was given a class IIa, level of evidence B-R recommendation.
Sacubitril/valsartan
ARNI (sacubitril/valsartan; or LCZ696) is a combination drug of neprilysin inhibitor and valsartan, and is indicated for use in symptomatic heart failure patients with reduced ejection fraction (HFrEF) with ejection fraction (EF) ≤35 %. (In the early days of the trial, the EF cutoff was ≤40 % and revised later to the current ≤35 %.) The ARNI trial (Prospective Comparison of ARNI with Angiotensin Converting Enzyme Inhibitors to Determine Impact on Global Mortality and Morbidity in Heart Failure; PARADIGM-HF) was published in September 2014.3 The trial was stopped early at 27 months of a projected 4-year duration. In light of the dramatic benefit with use of ARNI, the data safety and monitoring board decided it would be unethical to continue the trial with patients in the standard-of-care arm. The primary objective of PARADIGM-HF was to determine whether Entresto was superior to a renin-angiotensin system inhibitor (enalapril) alone in reducing the risk of the combined endpoint of cardiovascular (CV) death or hospitalization for heart failure. After discontinuing their existing angiotensin-converting enzyme (ACE) inhibitor or angiotensin II receptor blocker (ARB) therapy, patients entered sequential single-blind run-in periods during which they received enalapril 10 mg twice daily, followed by Entresto 100 mg twice daily, increasing to 200 mg twice daily. It is important to note the dropout rate was equal in the two treatment groups, reflecting that no matter how effective a medication, there will always be a percentage of the patient population intolerant to target doses.
Patients who successfully completed the sequential run-in periods were randomized to receive either Entresto 200 mg (n=4,209) twice daily or enalapril 10 mg (n=4,233) twice daily. The median follow-up duration was 27 months and patients were treated for up to 4.3 years. PARADIGMHF was the largest trial in heart failure history, screening over 10,000 patients and ultimately randomizing 8,442 patients to the gold standard ACE inhibitor as the control arm versus ARNI. Enalapril was chosen to represent the ACE category because it is the most studied drug in heart failure trials. An ACE inhibitor, and not ARB, was chosen in the standardof- care arm because the data for ACE inhibitors are stronger, and ACE inhibitors are still considered first-line therapy over ARBs for heart failure. The primary endpoint was CV death or hospitalization for heart failure. Entresto had a 20 % incremental reduction in CV death beyond that achieved with enalapril and an absolute risk reduction of 4.7 %.
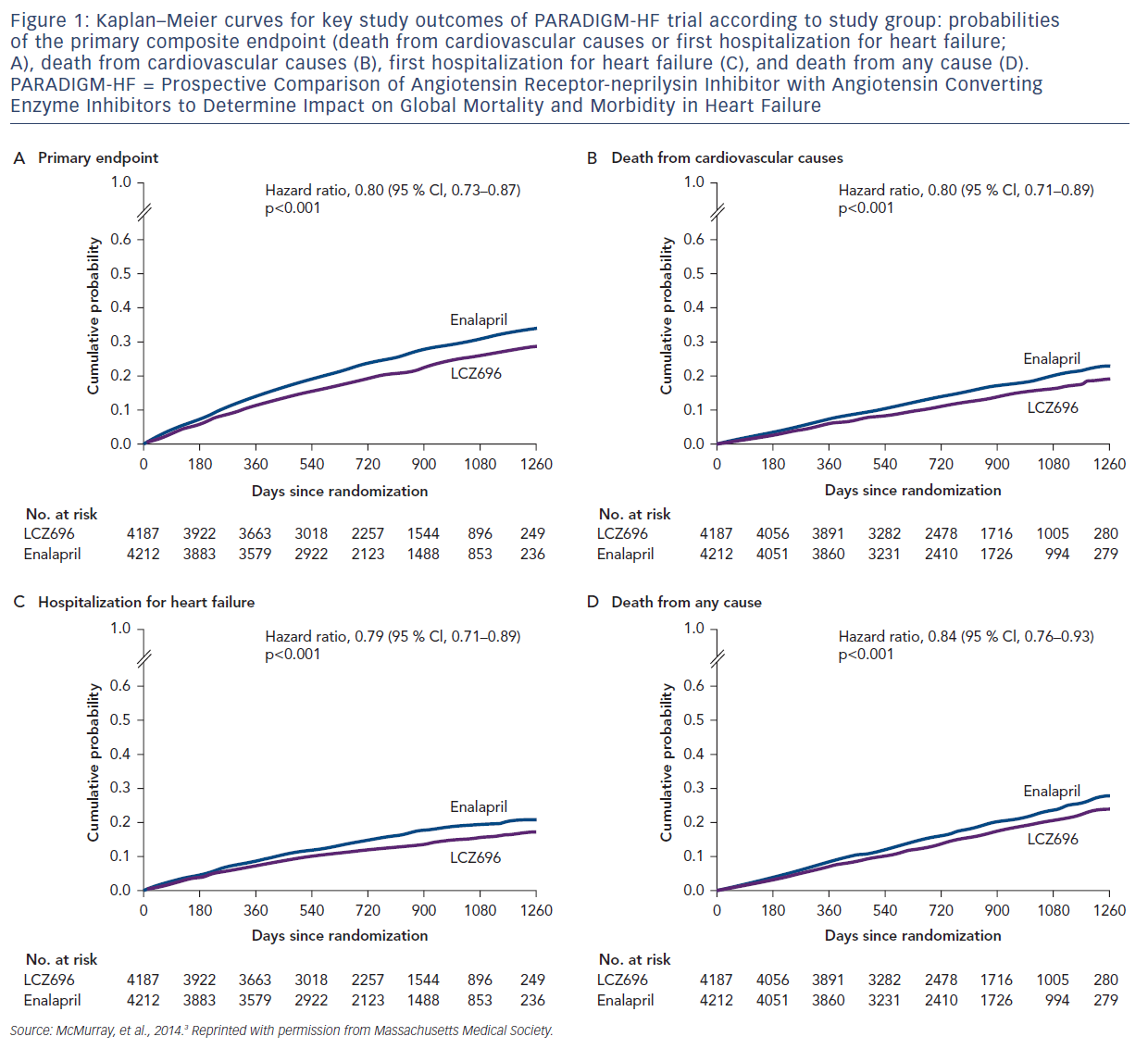
Figure 1 presents an overview of the key results in the original article.3 Using patient-level data, the PARADIGM-HF investigators estimated that treatment with sacubitril/valsartan prolongs life by an average of 1–2 years. The aggressive uptitration of dosages is reflected in a moderate percentage of patients withdrawing from the trial because of intolerance to the drugs. More patients dropped off in the enalapril arm (12.2 %) than in the ARNI arm (10.7 %). Dosing in the trial was based on the total amount of both components of sacubitril/valsartan. The PARADIGM-HF trial demonstrated that Entresto was superior to enalapril in reducing the risk of the combined endpoint of CV death or hospitalization for heart failure, based on a time-to-event analysis (p<0.0001). The treatment effect reflected a reduction in both CV death and heart failure hospitalization. Sudden death accounted for 45 % of CV deaths, followed by pump failure, which accounted for 26 %. Entresto also improved overall survival (p=0.0009). However, patients with severe HF were underrepresented in the PARADIGM-HF trial, as were African American patients, common in most first trials for heart failure. Also, the use of sacubitril alone for the treatment of HFrEF has not been evaluated.4
A recent analysis presented at this year’s HFSA Annual Scientific Meeting has now shown improved quality of life in the ARNI LCZ696 group over the enalapril group, especially in the overall Kansas City Cardiomyopathy Questionnaire clinical score.5
Concerns have been raised about neprilysin inhibition in regard to LCZ696. Neprilysin inhibition in mice does result in elevation of amyloidbeta peptide and plaque-like deposits, which are 30–50 times as high as normal levels in the brain.6 Cognition, memory, and dementia-related adverse events were not increased in the LCZ696 group in PARADIGM-HF. Indeed, it is possible that cognitive decline related to vascular disease might be reduced by LCZ696. A trial of LCZ696 versus valsartan, which includes repeated measurements of cognitive function in patients who have heart failure and a preserved EF, is ongoing (Efficacy and Safety of LCZ696 Compared to Valsartan, on Morbidity and Mortality in Heart Failure Patients With Preserved Ejection Fraction (PARAGON-HF), ClinicalTrials identifier NCT01920711). Neprilysin also inhibits prostate cancer cell invasion in vitro,7 and neprilysin overexpression has been associated with improved disease-free survival among women with breast cancer.8 Protection derives from the inactivation of mitogenic peptides, including endothelin-1 and bradykinin. No increase in the risk of cancer was associated with LCZ696, and 2-year carcinogenicity studies involving rodents that received the neprilysin inhibitor component of LCZ696 did not show an increase in the incidence of tumors.
Entresto is supplied as a tablet for oral administration. The recommended starting dose is 100 mg (49 mg sacubitril/51 mg valsartan) twice daily. Similar to current clinical practice with ACE and ARB uptitration, the dose of Entresto can be increased every 2–4 weeks with the objective to reach the target maintenance dose of 97/103 mg twice daily, as tolerated by the patient. While most patients will tolerate a simple doubling of dose, some will be more sensitive and require starting at the lower 24/26 mg twice daily dose combined with smaller increments and/or longer time periods between dose-change titration. This is important for patients not currently taking an ACE inhibitor or an ARB, who have lower systolic blood pressure or who have moderate renal or hepatic dysfunction.
Entresto is a combination of sacubitril, a neprilysin inhibitor, and valsartan, an ARB drug. Entresto inhibits neprilysin (neutral endopeptidase) via LBQ657, the active metabolite of the prodrug sacubitril, and blocks the angiotensin II type-1 (AT1) receptor via valsartan. Neprilysin inhibition leads to decreased metabolism of endogenous vasoactive peptides (e.g. natriuretic peptides, adrenomedullin, bradykinin, substance P, and calcitonin gene-related peptide).9–12 This inhibition produces elevated levels of these endogenous vasoactive peptides, and thus augmentation of their beneficial effects, including a decrease in adverse neurohormonal activation, lower vascular tone, decreased cardiac fibrosis, less hypertrophy, and decreased sodium retention. The CV and renal effects of Entresto in heart failure patients are attributed to the increased levels of peptides that are degraded by neprilysin, such as natriuretic peptides (by LBQ657) and the simultaneous inhibition of the effects of angiotensin II (by valsartan). Valsartan inhibits the effects of angiotensin II by selectively blocking the AT1 receptor, and also inhibits angiotensin II-dependent aldosterone release. The FDA approval of Entresto was based on the impressive results of the PARADIGM-HF trial for patients with symptomatic chronic heart failure (New York Heart Association [NYHA] class II–IV) and left ventricular EF ≤40 %.
Ivabradine
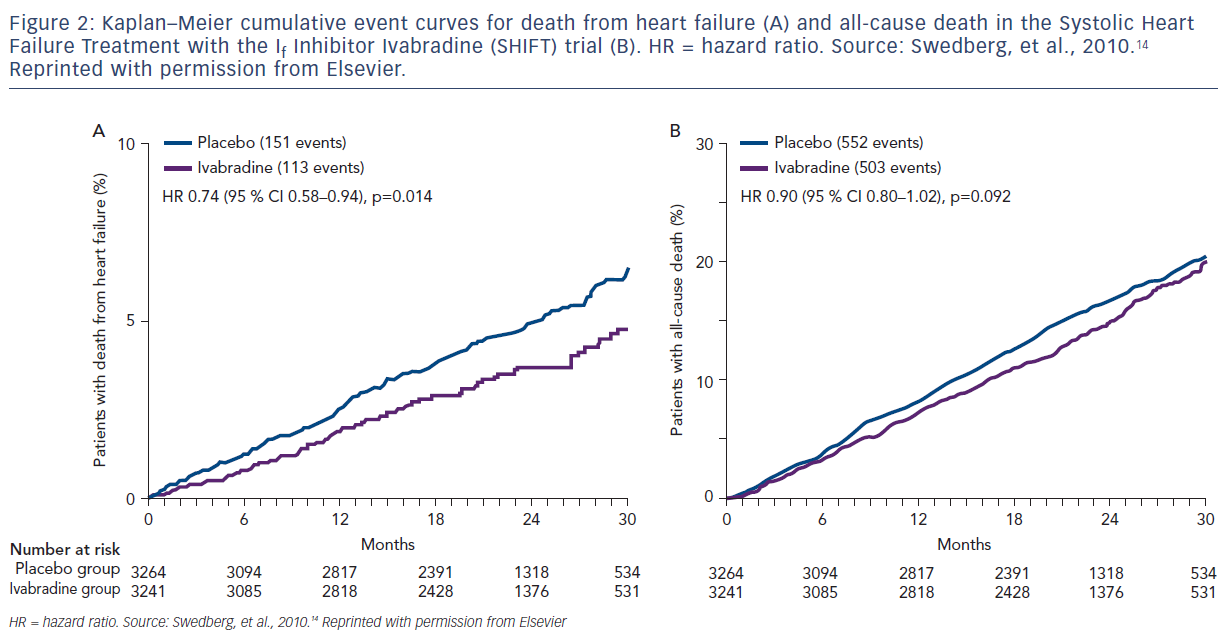
Ivabradine is the first drug of its kind with a novel mechanism of action. It selectively inhibits the pacemaker ‘funny’ (lf) channel, which is responsible for the autonomic capacity of the sinoatrial node. In patients with heart failure, If channels are upregulated in the atrial tissue.13 Ivabradine decreases the heart rate by direct sinus node inhibition without affecting blood pressure, myocardial contractility, or intracardiac conduction. The drug is approved for patients with stable, symptomatic, chronic heart failure with EF ≤35 % who are in sinus rhythm and have a resting heart rate >70 BPM, and are also taking beta-blockers at the highest tolerated doses. Ivabradine was studied in an industrysponsored clinical trial (Amgen) of 6,505 participants. The Systolic Heart Failure Treatment with the If Inhibitor Ivabradine (SHIFT) trial was a multinational, randomized, double-blinded, placebo-controlled trial published in 2010, which showed that ivabradine significantly reduced the primary composite endpoint of CV death and hospitalization for worsening heart failure by as much as 18 % (Figure 2).14 The finding was mainly attributable to ivabradine’s favorable effect on heart failure hospitalizations (16 % versus 21 %; hazard ratio [HR] 0.74, 0.66–0.83; p<0.0001) and less to heart failure deaths (3 % versus 5 %; HR 0.74, 0.58–0.94; p=0.014). Note, that CV deaths and all-cause mortality were not affected by ivabradine.
In the SHIFT trial, only 26 % of subjects were on target doses of betablocker and only 56 % of patients were on at least half-target doses of beta-blocker. Although this is presumably due to beta-blocker intolerance, an element of beta-blocker underuse may have overestimated the benefit of ivabradine. The effects of ivabradine were attenuated in a group of patients on at least 50 % target dose of beta-blocker, raising the question of whether the benefit of ivabradine may be limited to patients with complete or partial beta-blocker intolerance.
Corlanor is supplied as tablets for oral administration. The recommended starting dose is 5 mg twice daily. After 2 weeks of treatment the dose should be adjusted based on heart rate, with a maximum dose of 7.5 mg twice daily. In patients with conduction defects, or in whom bradycardia could lead to hemodynamic compromise, the initiate dosing is 2.5 mg twice daily. Part of the challenge, as well described in recent publication by Bhatt et al.15 is how to determine “maximal beta-blocker tolerated” prior to initiation of ivabradine.
Discussion
Amgen’s Corlanor and Novartis’ Entresto are both twice-a-day pills that currently cost approximately $4,500 per year, with Medicare and most private insurers covering the drugs. A new study published in Annals of Internal Medicine concluded these drugs are cost-effective in patients with NYHA class II–IV heart failure at a cost of $47,000 per quality-adjusted life year (QALY).16 Using a hypothetical cohort based on the characteristics of patients in the PARADIGM-HF trial, the costeffectiveness and cost per QALY gained of sacubitril/valsartan relative to enalapril was estimated.17 The average medication and heart failure hospitalization costs for patients in the sacubitril/valsartan and enalapril groups were $60,391 and $21,758, respectively. The use of sacubitril/ valsartan yielded 6.59 QALYs (9.48 life years), while enalapril yielded 5.83 QALYs (8.40 life years). This resulted in an incremental cost-effectiveness ratio for sacubitril/valsartan versus enalapril of $50,959 per QALY gained. Recent further analysis released at HFSA18 demonstrated the costeffectiveness is continuing to improve for sacubitril/valsartan to under $50,000 per life year gained and under $18,000 for ivabridine per life year gained, both being well within the range of what is considered acceptable financial cost.
A continual theme in the medical community is the underutilization of guideline-directed medical therapy; high costs and the need for more studies are often cited as the reason for not using optimum medications. Unfortunately, simply starting a medication is regarded as adequate. Advancement of effective medications takes time and effort which can be difficult in today’s reality of too many patients and too little provider time. Appropriate infrastructures need to be built so patients can be effectively treated in the ways identified and demonstrated by science as most successful.
Not all new drugs have been successful. RLX030 (serelaxin) in patients with acute heart failure (AHF) was studied in the Efficacy, Safety and Tolerability of Serelaxin When Added to Standard Therapy in AHF (RELAX-AHF-2) trial,19 but did not meet the primary endpoints of reduction in CV death through day 180 or reduced worsening heart failure through day 5 when added to standard therapy in patients with AHF. RLX030, a relaxin receptor agonist, is a recombinant form of the naturally-occurring human relaxin-2 hormone. Human relaxin-2 is present in both men and women; elevated levels in pregnant women are thought to help the body cope with the additional CV demands during pregnancy.
There is enthusiasm for future heart failure therapies, including omecamtiv mecarbil, a novel cardiac myosin activator. Cardiac myosin is the cytoskeletal motor protein in the cardiac muscle cell that is directly responsible for converting chemical energy into the mechanical force resulting in cardiac contraction. Cardiac myosin activators are thought to accelerate the rate-limiting step of the myosin enzymatic cycle and shift the enzymatic cycle in favor of the force-producing state. Preclinical research has shown that cardiac myosin activators increase contractility in the absence of changes in intracellular calcium in cardiac myocytes. The Chronic Oral Study of Myosin Activation to Increase Contractility in Heart Failure (COSMIC-HF) trial was a double-blind, randomized, placebo-controlled, multicenter, phase II trial designed to evaluate an oral formulation of omecamtiv mecarbil in chronic HFrEF.20 In the study, 448 chronic HFrEF were dosed with the selected oral formulation of omecamtiv mecarbil for 20 weeks and followed for a total of 24 weeks. Patients were randomized 1:1:1 to receive either placebo or treatment with omecamtiv mecarbil 25 mg twice daily or 25 mg with dose escalation to 50 mg twice daily, depending on plasma concentrations of omecamtiv mecarbil after 2 weeks of treatment. The primary endpoints for the expansion phase were to assess the maximum and predose plasma concentration of omecamtiv mecarbil. The secondary endpoints were to assess changes from baseline in systolic ejection time, stroke volume, left ventricular end-systolic diameter, left ventricular end-diastolic diameter, heart rate, and N-terminal pro-brain natriuretic peptide (NT-proBNP; a biomarker associated with the severity of heart failure) at week 20, as well as the safety and tolerability of omecamtiv mecarbil, including incidence of adverse events from baseline to week 24. Following 20 weeks of treatment, statistically significant improvements were observed in all prespecified secondary endpoint measures of cardiac function in the dose titration group, compared with placebo. Systolic ejection time increased by 25.0 ms (p<0.0001), stroke volume increased by 3.6 ml (p=0.0217), and heart rate decreased by 3.0 BPM (p=0.0070). Left ventricular end-systolic and end-diastolic dimensions decreased by 1.8 mm (p=0.0027) and 1.3 mm (p=0.0128), respectively, and were associated with statistically significant reductions in left ventricular endsystolic and end-diastolic volumes. NT-proBNP decreased by 970 pg/ ml (p=0.0069). In prespecified exploratory analyses of the dose titration group, placebo-corrected reductions in NT-proBNP persisted 4 weeks after stopping omecamtiv mecarbil, decreasing further to 1,306 pg/ml (p=0.0006). The data also showed increases in fractional shortening at week 20 compared with placebo in the dose titration group. COSMICHF, conducted by Amgen in collaboration with Cytokinetics, was not designed to assess the impact of omecamtiv mecarbil on CV outcomes in heart failure patients. New trials to assess clinical efficacy are underway.
Changing a clinical paradigm takes time, and the clinical community must retain a desire to always be open for new discoveries. If we look back at one of the greatest breakthroughs in heart failure medicine, betablockers, we are reminded that what we think we know is not always true. When beta-blockers were introduced, as now with these new drugs, there was resistance to changing patients’ medication profiles. Who remembers the days of office visits with vital sign checks every 15 minutes to make sure the patient would be okay with carvedilol 6.25 mg dosage? If this pattern was the directive today, would we even have explored beta-blockers further? Can you imagine a heart failure patient today not being offered beta-blockers? Change is difficult, but we must remember that we clinicians are only as good as our next discovery. We must embrace the new, respect that prior truths are not always forever, and offer our patients the best we have in the moment.