










Hypercholesterolemia is a central risk factor for the development of atherosclerotic cardiovascular disease (ASCVD), the leading cause of death and disability worldwide. Although the increased utilization of lipid-lowering therapy has decreased the mean LDL cholesterol (LDL-C) level from 3.26 mmol/l (126 mg/dl) in 1999–2000 to 2.88 mmol/l (111 mg/dl) in 2013–2014, recent observations suggest that long-term exposure to even modestly elevated plasma LDL-C concentrations is associated with a greater risk of coronary heart disease (CHD) later in life.1.2
Moreover, many individuals cannot achieve adequate LDL-C reduction with standard lipid-lowering therapies or are unable tolerate them, specifically statins, at the optimal dose. In addition, individuals with familial hypercholesterolemia (FH), the single most common Mendelian disorder with an estimated prevalence of around one in 250, are exposed to severe hypercholesterolemia from birth and are at considerably higher risk for premature ASCVD.3 As with patients with established ASCVD, those with FH frequently require the addition of non-statin therapies, such as ezetimibe or proprotein convertase subtilisin kexin 9 (PCSK9) inhibitors.
In 2003, a missense mutation causing gain-of-function of PCSK9 was discovered as the third genetic locus for FH.4 This discovery ushered in intense research efforts that, ultimately, elucidated the mechanism by which gain-of-function of PCSK9 leads to high plasma LDL-C.
PCSK9 is secreted into the plasma primarily by hepatocytes at low concentrations where it then acts as a second ligand for the LDL receptor (LDLR). Specifically, PCSK9 binds LDLR on the hepatocyte cell surface as the LDLR enters the cell in a ternary complex with PCSK9 and LDL bound to it. Ordinarily, after receptor-mediated endocytosis, the internalized LDL–LDLR complex decouples when the internal compartment of the endosome reaches a critical pH with LDLR recycling back to the surface. However, when PCSK9 is bound to the LDLR, it interferes with the natural LDLR recycling loop and the entire ternary complex (LDL–LDLR–PCSK9) stays within the endosome and fuses with the lysosome where the constituent components are digested.5
As gain-of-function mutations are relatively rare, investigators sought to determine if naturally occurring loss-of-function (LOF) mutations in the PCSK9 gene existed as well. Indeed, several LOF mutations were discovered. These LOF mutations in PCSK9 were associated with lifelong reductions, albeit generally modest, in plasma LDL-C levels and marked reductions in cardiovascular risk.6–9
These seminal observations served as proof in principle that antagonizing PCSK9 may have significant therapeutic potential. What follows is an overview of the various therapeutic classes that inhibit PCSK9, their associated clinical trial data, and future directions.
Therapeutic Monoclonal Antibodies
The development of the therapeutic anti-PCSK9 monoclonal antibodies (mAbs) serves as perhaps the best example of how genetic insights can facilitate targeted drug development. It took 12 years from the discovery of PCSK9 as a low-abundance circulating protein with an outsized impact on LDL metabolism to the regulatory approval of a therapy that antagonizes its action by the US Food and Drug Administration (FDA).

PCSK9 inhibitors were initially approved by the FDA on the basis of their LDL-C lowering efficacy and safety while the large cardiovascular outcomes trials were ongoing. Both therapeutic antibodies are fully human, target the same region of PCSK9, and have similar LDL-C lowering efficacy (a reduction of about 60% in LDL-C) at maximum doses.10,11 While they possess some structural differences (alirocumab is an IgG1 mAb and evolocumab is an IgG2 mAb), it is unclear whether this has any clinical significance. Thus far, the results of each of the large cardiovascular outcomes trials were similar between the two drugs (Table 1).
The FOURIER trial was the first of the randomized controlled outcomes trials to be reported.10 FOURIER enrolled 27,564 patients with atherosclerotic cardiovascular disease and LDL-C levels >1.81 mmol/l (>70 mg/dl) on optimized statin therapy. Patients were randomly assigned to receive subcutaneous injections of evolocumab (either 140 mg every 2 weeks or 420 mg monthly) or placebo. The primary endpoint was the composite of cardiovascular death, MI, stroke, hospitalization for unstable angina, or coronary revascularization (major adverse cardiac events, MACE). At 48 weeks, evolocumab therapy, compared to placebo, resulted in a 59% reduction in LDL-C. Evolocumab significantly reduced the risk of the primary endpoint compared to placebo (9.8% versus 11.3%; HR 0.85; 95% CI [0.79–0.92]; p<0.001). There were no significant differences between evolocumab and placebo with regard to adverse events including new-onset diabetes and neurocognitive events. However, evolocumab was associated with a higher incidence of injection-site reactions.10
The following year, the ODYSSEY OUTCOMES trial was published.11 This was a randomized, double-blind, placebo-controlled trial of 18,924 subjects with recent acute coronary syndrome (within 1–12 months before randomization) that compared the effect of alirocumab subcutaneously in a 75 mg dose versus placebo on incident MACE. Most trial participants (about 90%) received treatment with high-intensity statin therapy and had one or more of the following: LDL-C 1.81 mmol/l≥ (70 mg/dl); non-HDL cholesterol ≥2.60 mmol/l≥ (100 mg/dl); or apolipoprotein B ≥ 2.07 mmol/l (80 mg/dl). The primary endpoint was time to first occurrence of a 4-point composite MACE outcome (the trial did not include revascularization as in FOURIER).12
In the intention-to-treat analysis, alirocumab therapy, compared to placebo, resulted in 62.7%, 61.0%, and 54.7% reductions in LDL-C at 4 months, 12 months, and 48 months, respectively. The primary endpoint occurred in 9.5% of subjects allocated to alirocumab and in 11.1% of subjects allocated to placebo (HR 0.85; 95% CI [0.78–0.93]; p<0.001). The incidence of adverse events and laboratory abnormalities was similar in the alirocumab and placebo groups with the exception of injection-site reactions (3.8% in alirocumab versus 2.1% in placebo; p<0.001). These injection-site reactions included itching, redness, and swelling and were generally mild and self-limiting.11
One of the interesting and unanticipated facets of PCSK9 inhibition with therapeutic mAbs is the association with plasma lipoprotein (a) (Lp(a)) lowering (Table 2). A recent meta-analysis that assessed 27 randomized controlled clinical trials enrolling 11,864 subjects demonstrated comparable reductions in Lp(a) associated with PCSK9 inhibitor treatment (Lp(a) reduction of 21.9% (95% CI [−24.3 to −19.5]).13 Given the epidemiological and genetic associations of Lp(a) with ASCVD, there is interest in evaluating the potential role of PCSK9 inhibitor-induced reduction in Lp(a) to reduce ASCVD events.14
Recent analyses from the FOURIER and ODYSSEY OUTCOMES trials lend credence to the notion that reductions in Lp(a) via PCSK9 inhibition may effectively reduce residual ASCVD risk.15 O’Donoghue et al. evaluated plasma Lp(a) concentration in the FOURIER participants and found that higher baseline Lp(a) concentration associated with an increased rate of cardiovascular events independent of LDL-C concentration.16 Evolocumab was associated with an Lp(a) reduction of 27% at 48 weeks.16 Additionally, the use of evolocumab was associated with greater cardiovascular benefit in individuals with a higher than median baseline Lp(a) than in participants with a below the median baseline Lp(a) (23% reduction; HR 0.77; 95% CI [0.67–0.88]) compared to a reduction of 7% (HR 0.93; 95% CI [0.80–1.08]) in cardiovascular events respectively.16
The ODYSSEY OUTCOMES investigators extended these findings to models of absolute change in Lp(a) as a predictor of MACE. They found, in a fully adjusted model, that for each 0.026 mmol/l (1 mg/dl) lowering of Lp(a) associated with alirocumab, the corresponding event reduction was 0.6%. However, when modeling to more significant Lp(a) lowering, they were able to demonstrate significant relative risk reductions attributable to Lp(a) lowering. For example, absolute reductions in Lp(a) of 0.52 mmol/l (20 mg/dl) and 1.30 mmol/l (50 mg/dl) would be associated with relative risk reductions in MACE of 11% and 26%, respectively.
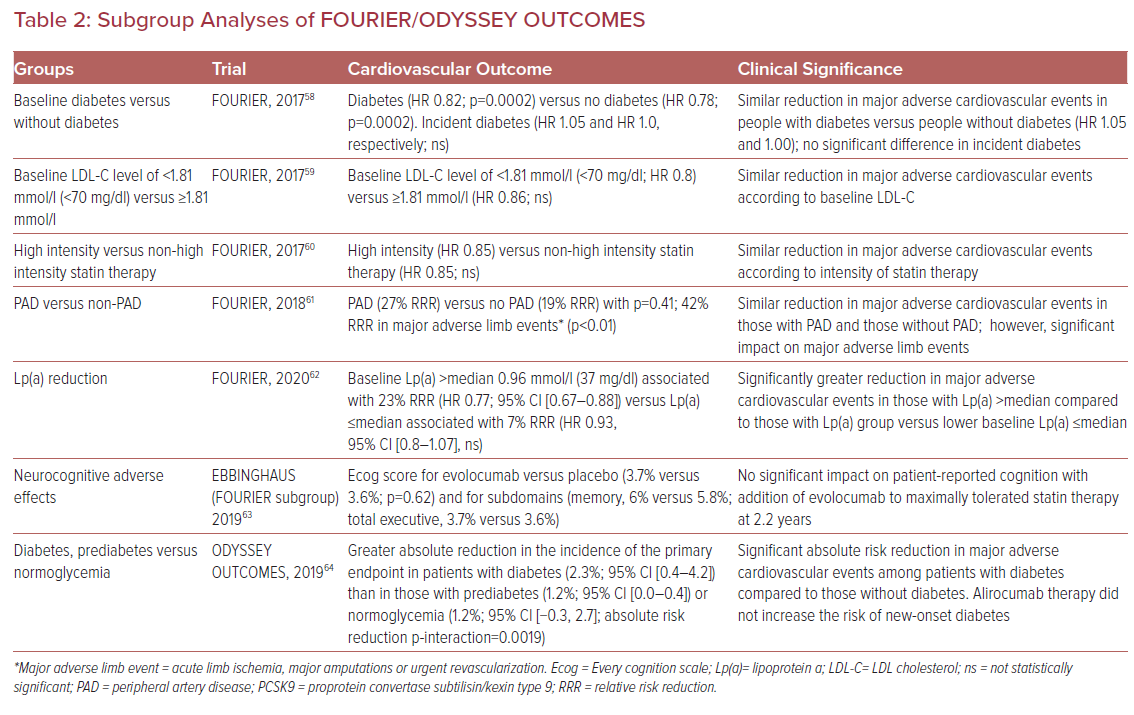
Importantly, the proportion of MACE reduction attributable to changes in Lp(a) and corrected LDL-C become substantial in those with the highest baseline levels of Lp(a) (greater than the 75th percentile of the population).17 The findings from the subanalyses of FOURIER and ODYSSEY OUTCOMES with respect to Lp(a) lowering are interesting and beg the question as to the potential future role of PCSK9 inhibitors for use in individuals with ASCVD and elevated Lp(a), irrespective of LDL-C.
At this point, the mechanisms by which PCSK9 inhibition is associated with reductions in plasma Lp(a) remains unclear. The most commonly touted hypothesis asserts that there is increased clearance of Lp(a) particles through the LDLR pathway. However, the idea that clearance of Lp(a) is facilitated by the LDLR presents a number of challenges:
- The affinity of Lp(a) for the LDLR is considerably less than that of LDL.18
- Lp(a) catabolic rates are similar in FH and non-FH patients.19
- Statins, which upregulate LDLR, are not associated with reductions in plasma Lp(a) concentration.20
Recent data suggest the possibility of alternative pathways beyond LDLR-mediated clearance involved in Lp(a) reduction by PCSK9 inhibition.21,22 In these clinical studies, the investigators found a significant discordance between LDL-C and Lp(a) lowering with PCSK9 inhibition, with a significant proportion of patients demonstrating essentially no meaningful Lp(a) lowering in the face of an excellent LDL-C response to therapy. Given that PCSK9 and, by extension, PCSK9 inhibitors affect a myriad of receptors beyond LDLR (e.g. APOER2, LRP1, VLDLR, CD36, and TLR2 plasminogen receptors), it is conceivable that there is an Lp(a) receptor, which is, at least in part, under the regulation of PCSK9. The LDL-C/Lp(a) discordance observed in the studies described above may be because Lp(a) clearance is mediated by apolipoprotein(a) isoform size.
It is also important to consider recent human metabolic studies that address the issue of PCSK9 inhibition and Lp(a) lowering. In general, these demonstrate that PCSK9 inhibitors reduce plasma Lp(a) concentrations by accelerating the catabolism of Lp(a) particles without changing the apolipoprotein(a) production rate. This observation may potentially be explained by the extreme upregulation of hepatic LDLR and/or reduced competition between LDL and Lp(a) particles for LDLR once PCSK9 inhibition has dramatically reduced plasma LDL-C.23,24
Silencing RNA
The potential use of silencing RNA (siRNA) as a therapeutic tool to inhibit PCSK9 could be a new frontier in LDL-C management. Inclisiran is the siRNA therapeutic furthest along in development. While mAbs solely target circulating PCSK9 (extracellularly), inclisiran targets intracellular PCSK9 by inhibiting the translation of PCSK9 messenger RNA (mRNA). Therapeutic siRNAs are short sequences of double-stranded RNA that enter the cell and bind to the RNA-induced silencing complex, allowing it to cleave the mRNA, in this case coding for PCSK9.25 Inclisiran is a long-acting synthetic siRNA bound to a carbohydrate (multivalent N-acetylgalactosamine) for which the liver expresses abundant receptors, allowing hepatocyte targeting.26,27 This targeted approach means that lower doses can be used to achieve the desired effect, which decreases the risk of adverse effects.
In 2014, Fitzgerald et al. performed a single-blind, placebo-controlled phase I trial of 32 healthy volunteers randomized to receive a dose of ALN-PCS (the precursor to inclisiran) in various doses versus placebo. Inclisiran at a maximum dose of 400 mg/kg reduced circulating PCSK9 concentration by 70% and reduced mean LDL-C level by 40% from baseline at 6 months compared to placebo. At lower doses of 250 mg/kg and 150 mg/kg, reductions in circulating PCSK9 plasma protein were 70% and 65%, respectively, and reductions in LDL-C level at 6 months were 28% and 22%, respectively, compared to placebo. The most common adverse events were cough, musculoskeletal pain, nasopharyngitis, headache, back pain, and diarrhea. Further research into the safety of ALN-PCS revealed no drug-related serious adverse events in a cohort of 24 participants.25
A follow-up phase I trial by Fitzgerald et al. also assessed the efficacy of a single-dose phase versus a multiple-dose phase. In the single-dose phase (25 mg, 100 mg, 300 mg, 500 mg, and 800 mg of inclisiran), inclisiran at a dose of 300 mg or higher was associated with significant reductions in plasma PCSK9 concentration compared to placebo, with similar magnitudes of reduction across the 300–800 mg does range (least-squares mean change: 69.9–74.5%). The largest reduction in plasma PCSK9 concentration was 74.5% and was observed in the group treated with 300 mg of inclisiran.
With respect to LDL-C levels, significant reductions from baseline were observed from doses of 100 mg and above, with the largest reduction of 50.7% in LDL-C occurring with the 500 mg dose. These reductions were sustained at 6 months with doses greater than 300 mg, but were not observed with the 25 mg and 100 mg doses. The multiple-dose phase (125 mg weekly for four doses, 250 mg every other week for two doses, and 300 or 500 mg monthly for two doses) demonstrated the greatest reduction in plasma PCSK9 (84.7%) with the 500 mg monthly for two doses, and the greatest reduction of LDL-C (60%) with the 300 mg monthly for two doses.28
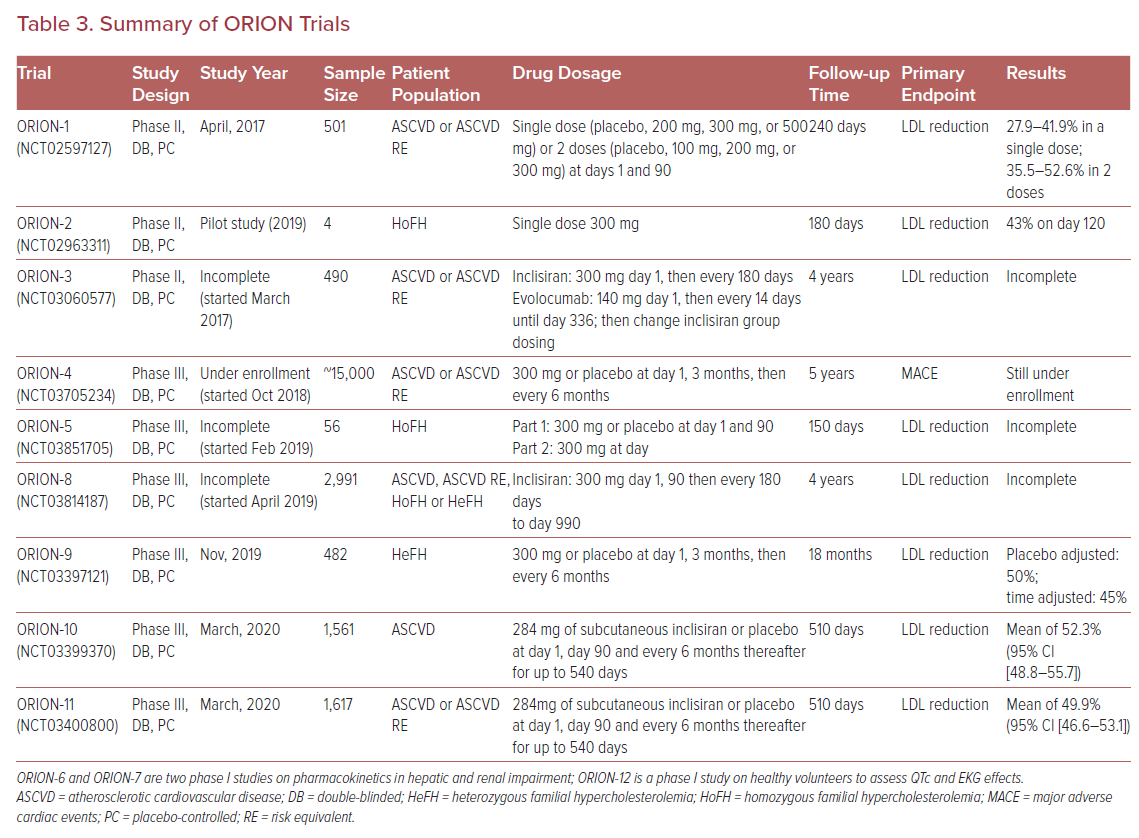
Ray et al. followed-up the initial studies with ORION-1, a phase II multicenter, double-blinded, randomized controlled trial of 501 subjects. The trial assessed the LDL-C lowering efficacy of inclisiran at 180 days as the primary endpoint. Trial participants received a single dose of placebo, 200 mg, 300 mg, or 500 mg of inclisiran or two doses of placebo, 100 mg, 200 mg, or 300 mg of inclisiran at days 1 and 90. The LDL-C reduction ranged from 27.9-41.9% in subjects who received a single dose of inclisiran and 35.5–52.6% in subjects who received two doses. Two doses of inclisiran at 300 mg was the regimen associated with the greatest LDL-C lowering efficacy with percentage and mean reductions of LDL-C levels of 52.6% and 1.66 mmol/l (64.2 mg/dl), respectively, at 180 days, and 47.2% and 1.53 mmol/l (58.9 mg/dl) respectively at 240 days. Regardless of dosing regimen, the lipid-lowering effects of inclisiran were sustained at day 240. The adverse effect profile was similar to that previously described. Importantly, the incidence of adverse events did not differ significantly between the inclisiran and placebo groups.29
Recently, two phase III randomized placebo-controlled trials were published, including the ORION-10 trial that enrolled 1,561 patients with ASCVD, and the ORION-11 trial that enrolled 1,617 patients with ASCVD or an ASCVD risk equivalent. All patients in both trials had elevated LDL-C despite maximal statin therapy, and were randomized to receive either 284 mg of subcutaneous inclisiran or placebo at day 1, day 90, and every 6 months thereafter for up to 540 days. At day 510, inclisiran reduced LDL-C levels by 52.3% (95% CI [48.8–55.7]) in the ORION-10 trial and by 49.9% (95% CI [46.6–53.1]) in the ORION-11 trial with p values <0.001 for all comparisons against placebo. All-cause adverse events were similar between the inclisiran and placebo groups (73.5% with inclisiran versus 74.8% with placebo in ORION-10 and 82.7% with inclisiran versus 81.5% with placebo in ORION-11), with the exception of injection-site reactions, which were higher in the inclisiran group (2.6% versus 0.9% in the ORION-10 and 4.7% versus 0.5% in the ORION-11); however, such reactions were generally mild.30
Inclisiran continues to be tested in special populations. It may be a promising strategy for LDL-C management in patients with severe chronic kidney disease, where the use of statins can be challenging due to issues related to clearance, tolerability, and adverse events.31 Analysis of ORION-7, a phase I single-dose non-randomized trial evaluating subjects with various degrees of renal impairment, demonstrated that the pharmacodynamics and safety profile of inclisiran were similar in participants with normal and impaired renal function (including patients with severe renal impairment, with an estimated glomerular filtration rate of <30 ml/min/1.73 m2).32 An analysis of ORION-1 demonstrated that inclisiran was equally effective in diabetic and nondiabetic populations, with median dose-dependent LDL-C reductions of 28–53%.33
Various other phase II and III trials within the ORION program are still under way to assess the long-term safety and efficacy of inclisiran (Table 3). ORION-3 is a phase II, open-label, non-randomized trial that compares inclisiran to evolocumab in patients with clinical ASCVD, an ASCVD risk equivalent, or heterozygous FH to assess the efficacy and safety profile of inclisiran for up to 4 years. ORION-4 is a large phase III cardiovascular outcomes trial that will assess MACE at 6 years or more. Other phase III trials, such as ORION-5 and ORION-9, will assess the efficacy of inclisiran in homozygous and heterozygous FH, respectively.34 Both trials showed significantly reduced LDL-C levels with inclisiran compared to placebo. In 482 heterozygous FH patients, at day, 510 LDL-C levels were reduced by 39.7% in the inclisiran group compared to an increase of 8.2% in placebo group (p<0.001).35 A smaller study of four subjects with homozygous FH on background high-intensity statin and ezetimibe therapy found that all participants achieved robust and durable PCSK9 reductions of 40–80% at day 180, as well as LDL-C reductions of 17–37% that were observed in three out of the four patients.36
Vaccines
Another promising approach to inhibit PCSK9 involves priming the immune system to neutralize PCSK9 using PCSK9-peptide-based vaccines. Several types of vaccines are in development. In 2017, Pan et al. used a virus-like particle (VLP)-peptide vaccine targeting PCSK9 in mouse models and found that the mice developed high titer anti-PCSK9 IgG antibodies that resulted in decreased plasma PCSK9 levels, upregulated LDLR expression in the liver, and decreased total plasma cholesterol with no detectable immune injury.37
Around the same time, Landlinger et al. developed an AT-04A anti-PCSK9 vaccine that elicited persistent elevations in anti-PCSK9 antibodies with a significant 53% reduction in total plasma cholesterol. Moreover, the treated mice exhibited reductions in LDL-C and various inflammatory markers (such as vascular endothelial growth factor). Of note, treatment with the AT-04A vaccine resulted in decreased atherosclerotic lesion area and aortic inflammation in intervention compared to control mice.38
A year later, Kawakami et al. tested a similar peptide vaccine consisting of short peptides conjugated to a carrier protein that was administered to ApoE-deficient mice and elicited a significant antibody response to PCSK9 with titers maintained at 24 weeks. These mice exhibited increased cell-surface LDLR expression and decreased total cholesterol, very LDL, and chylomicron concentration with effects sustained at 24 weeks.39
Another approach to vaccine development involves binding immunogenic peptides (consisting of fused PCSK9-tetanus [IFPT] linked to short PCSK9 peptides) to negatively charged nanoliposomes using DSPE-PEG-maleimide lipid (L-IFPT) and adsorbed to Alhydrogel (Croda) aluminum hydroxide gel, forming an L-IFPTA+ vaccine. In mouse models, this L-IFPTA+ vaccine induced an effective IgG antibody response with an associated 58.5% decrease in plasma PCSK9 compared to controls and decreased total cholesterol, LDL-C and VLDL-C by 44.7%, 51.7%, and 19.2%, respectively, at 8 weeks. At 16 weeks after vaccination, the LDL-C reduction was sustained and had decreased by 42% from baseline.40 Further analysis over 48 weeks demonstrated that the L-IFPTA+ vaccine was able to stimulate a long-lasting humoral immune response against the PCSK9 peptide and resulted in increased anti-inflammatory mediators, such as CD4+ Th2 cells and IL-4 within splenocytes. These findings suggest that this vaccine has the potential for long-term therapeutic cholesterol lowering.41
CRISPR
Genome editing, such as clustered regularly interspaced short palindromic repeats (CRISPR), can be leveraged as a potential therapeutic to disrupt target genes, such as PCSK9. CRISPR-associated systems (Cas) use the Cas9 (CRISPR-Cas9) nuclease derived from bacteria, such as Streptococcus pyogenes or Staphylococcus aureus, that binds to DNA and causes double-stranded breaks with the goal of introducing LOF mutations into the endogenous PCSK9 gene in vivo. Initial studies in animal models found that on-target mutagenesis rates in the liver were as high as >50% and associated with a reduction in plasma PCSK9 concentration, increased LDLR expression, and a 35–40% reduction in plasma cholesterol. Moreover, this approach demonstrated sustained PCSK9 reduction over 24 weeks.42 Importantly, no off-target mutagenesis was detected.43
Despite these positive results with CRISPR gene editing, safety concerns continue regarding the unpredictable nature of the cellular repair of double-stranded breaks, as well as off-target mutagenesis. New advancements favor the use of base editors comprised of CRISPR-Cas9 fused to a cytosine deaminase domain that modify cytosine to thymine bases (C–T changes) at precise locations in the genome without the need for double-strand DNA breaks or DNA replication. The advantage of this approach is a superior safety profile.44 In an adult mouse model, Chadwick et al. demonstrated that the use of a base editor targeting PCSK9 resulted in >50% reduction in plasma PCSK9 concentration and a >30% reduction in plasma cholesterol levels without evidence of off-target mutagenesis.45 Despite these advances, CRISPR technology is far from clinical application.
Other Methods of PCSK9 Inhibition
Additional approaches to antagonizing PCSK9 focus on inhibiting various steps in translation, post-translational processing, and binding of PCSK9 to the LDLR. Lintner et al. created an orally active compound (PF-06446846) that inhibits the translation of PCSK9 by inducing the ribosome to stall on codon 34, resulting in reduced plasma PCSK9 and total cholesterol levels in rats.46 However, one major limitation of this translational inhibitor is the lack of selectivity for PCSK9, which likely poses a challenge for future work in this area.47
Another approach to inhibiting PCSK9 production involves post-transcriptional downregulation of PCSK9 mRNA using miRNA mimetics. A recent study found that miR-191, miR-222, and miR-224 bind directly to the 3’ end of PCSK9 mRNA, thereby downregulating its expression into protein.48 However, these miRNA are again limited by the lack of selectivity to PCSK9.49
Downstream of translation, an emerging method targets the autocatalytic process of proPCSK9 (a zymogen) into active PCSK9. Active PCSK9 exits the endoplasmic reticulum (ER) only after proPCSK9 undergoes this autocatalytic cleavage process to generate a heterodimer of mature PCSK9.49,50 In certain families who are heterozygous for a PCSK9 LOF mutation, a resultant proPCSK9-Q152H variant was discovered, which caused proPCSK9 to be unable to undergo autocatalytic cleavage, thereby retaining the proPCSK9 variant within the ER and resulting in very low levels of circulating PCSK9 and serum LDL-C.51
Interestingly, after cotransfection of these proPCSK9-Q152H variants with wild-type PCSK9, it was observed that the variant dimerizes with wild-type PCSK9, forcing its retention within the ER and resulting in 78% less secretion of the wild-type PCSK9 protein.50,51 The development of a small molecule inhibitor to interfere with this autocatalytic process pose challenges since the zero-order kinetics of the autocatalytic process of proPCSK9 into PCSK9 requires high doses of an inhibitor to outcompete the intramolecular reaction. Additionally, it remains to be seen whether a small molecule that can traverse both the plasma and the ER membranes can be developed.52,53
There is also interest in developing a small molecule inhibitor that targets the PCSK9-LDLR interface. PCSK9 has an epidermal growth factor precursor homology domain A (EGF-A) binding site that interacts with the EGF-A domain of LDLR. The first EGF-A-like peptide, created in 2012, was an Fc fusion protein variant of EGF66 that bound to PCSK9 with inhibition of LDLR degradation both in vitro and in vivo mouse models.54
Several years later, the same team led by Zhang et al. developed a shorter peptide sequence of 13 amino acids, Pep2–8, that bound PCSK9 with greater affinity; however it was less active than its predecessor, EGF66.55 Furthermore, refinement of a small molecule inhibitor is difficult given the flat interface of the EGF-A binding site. However, Zhang et al. discovered a vacated N-terminal groove of PCSK9 adjacent to the EGF-A binding site that is accessible to small peptides, where a small molecule inhibitor can prevent the PCSK9–EGF-A interaction. Indeed, an engineered small molecule therapeutic peptide, MESFPGWNLV(hR)IGLLR, effectively inhibits PCSK9 binding to the EGF-A domain of LDLR.56 This recent structural identification of a novel pocket in PCSK9 opens up the door to future development of an orally active, small-molecule PCSK9 inhibitor.57
Conclusion
Over the past two decades, rapid, innovative developments have emerged within the field of therapeutic cholesterol lowering, especially in the area of PCSK9 inhibition. There are two approved PCSK9 inhibitors, evolocumab and alirocumab, with potent and equivalent LDL-C reductions of around 60%. Both drugs, when given in addition to standard of care, demonstrated improvement in cardiovascular outcomes in randomized controlled trials.
In addition to the PCSK9 mAbs, the development of siRNA as a means to inhibit PCSK9 has garnered much attention because of its LDL-C lowering efficacy, as well as its sustained durability after dosing. Several trials are still under way to assess the impact of inclisiran on cardiovascular outcomes and its efficacy in special populations.
Other innovative modes of PCSK9 inhibition that are still in the early phases of development include vaccines, CRISPR editing, and PCSK9 antagonists along various stages of translation, and small-molecule inhibitors that block the PCSK9-LDLR interaction. However, these novel agents are still in early development and further research will be required to demonstrate the future role of these therapies in the treatment of hypercholesterolemia and ASCVD.