










Heart failure is a major cause of hospital admissions, and it is associated with significant morbidity and mortality.1 Heart failure is expected to affect approximately 3.0% of US citizens by 2030.2 The diagnosis and treatment of new-onset systolic dysfunction can be challenging, particularly in patients presenting with cardiogenic shock. An often underrecognized culprit of acute heart failure is drug cardiotoxicity. We present a case of a man with paranoid schizophrenia who presented to a local hospital in undifferentiated shock and new systolic dysfunction of unknown etiology.
Case Presentation
A 37-year-old African-American man with a long-standing history of paranoid schizophrenia and multiple admissions for psychiatric hospitalization presented to the hospital with tachycardia, tachypnea, hypotension, and altered mental status requiring intubation upon arrival. A week earlier, the patient had been discharged following a 3-week psychiatric hospitalization for which he was started on clozapine and injectable fluphenazine. Regarding his social history, the patient smoked half a pack per day, and previously used cocaine and marijuana. He had no known family or past surgical history.
On admission, the patient tested negative for COVID-19. A chest CT scan revealed extensive bilateral infiltrates and was negative for pulmonary embolism. His initial elevated troponin levels were concerning for MI, so the patient was started on appropriate antithrombotic therapy. He was also started on broad-spectrum antibiotics to treat possible sepsis. He required vasopressors for refractory hypotension. An echocardiogram showed a global systolic dysfunction with reduced ejection fraction of 20–25%. Over the course of his hospitalization, the patient developed AF with rapid ventricular response requiring an amiodarone drip. Due to his worsening clinical condition requiring a higher level of care, the patient was transferred to our hospital’s coronary intensive care unit (CICU).
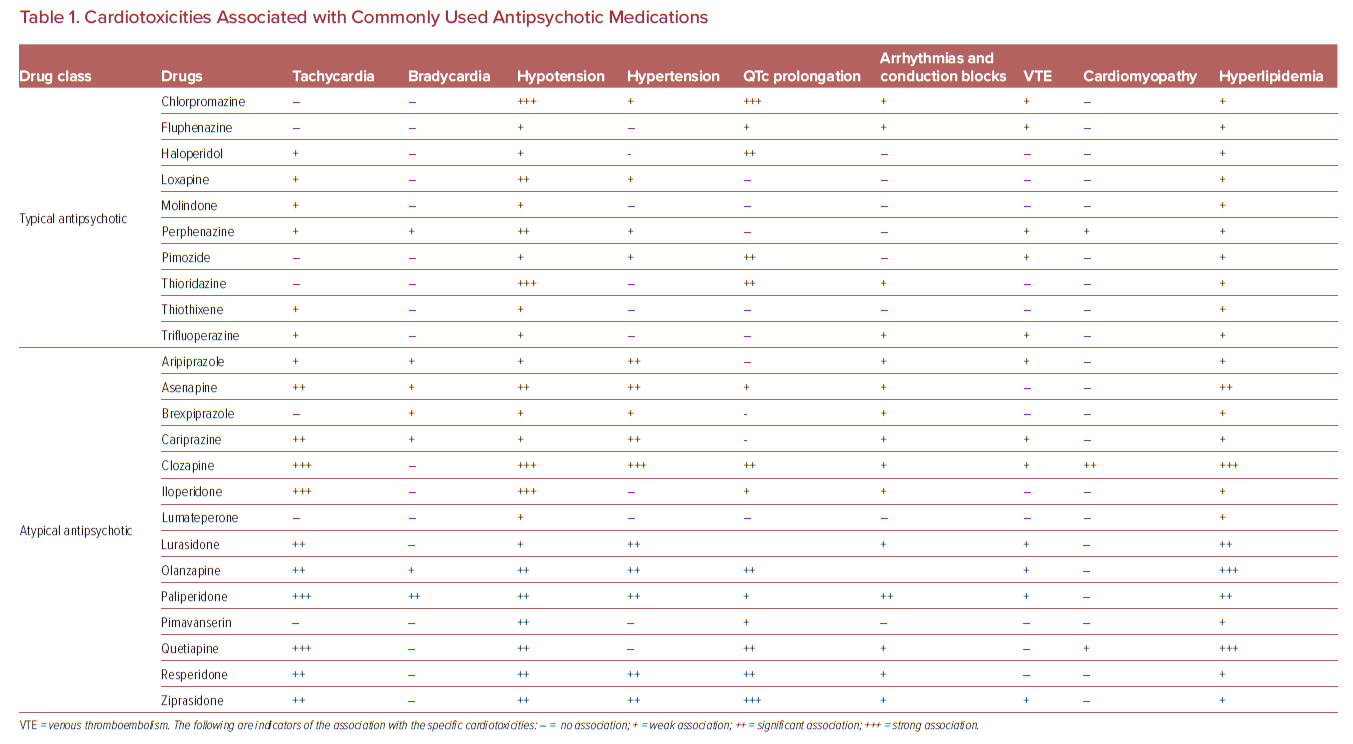
Upon arrival to the CICU, the patient was intubated (FiO2 of 40%), sedated, and hypotensive at 88/55 mmHg. On examination, he had sinus tachycardia without murmurs, rubs, or gallops, and had bilateral coarse breath sounds. The rest of his physical examination was unremarkable. Laboratory testing showed leukocytosis, hyponatremia, and hyperglycemia with signs of end-organ hypoperfusion, including acute kidney injury and elevated liver enzymes (Table 1). Importantly, his high-sensitivity troponin showed a downtrend from 2.60 ng/dl on presentation to the local hospital to 0.733 ng/ml at the CICU. A similar trend was observed with his lactate levels, which also declined from 3.2 to 1.8 IU/l. Earlier serologic tests at the local hospital for legionella, mycoplasma, streptococcus pneumonia, hepatitis B and C, and HIV were all negative. Furthermore, a chest X-ray showed bilateral opacities with marked cardiomegaly consistent with earlier imaging at the local hospital prior to transfer. The patient’s 12-lead ECG showed normal sinus rhythm without signs of new ischemic changes or pericardial pathologies (Figure 1). A bedside echocardiogram showed biventricular dysfunction without significant valvular abnormalities (Supplementary Material Video 1).
In the settings of undifferentiated shock, pulmonary artery catheterization may be a useful tool in distinguishing between cardiogenic and septic etiologies.3 This patient had an elevated central venous pressure of 21 mmHg, pulmonary arterial mean pressure of 35 mmHg, and pulmonary capillary wedge pressure of 26 mmHg, with a prominent decrease in cardiac index to 1.46 l/min and a mixed venous oxygen saturation of 50%, suggestive of a cardiogenic etiology of his shock. The patient was kept on broad-spectrum antibiotics to cover for a possible underlying infection complicating his shock.
Over the course of his CICU stay, the patient responded well to stabilizing therapies, including intravenous vasopressors for hypotension and diuretics for volume overload, with significant improvement in his hemodynamic status. To identify the etiology of his new systolic dysfunction, an ischemic evaluation was pursued with left heart catheterization, which showed angiographically non-obstructive coronary artery disease with normal Thrombolysis In Myocardial Infarction 3 flow. Hence, this patient was considered to have cardiogenic shock secondary to non-ischemic cardiomyopathy. To further evaluate this patient’s cardiomyopathy, a cardiac MRI study was suggested; however, the patient’s mechanical respiratory support precluded this evaluation. The patient was subsequently tested for viral and endocrine etiologies of non-ischemic cardiomyopathy; however, the workup was unrevealing. During that time, he was kept off his clozapine. Following hemodynamic stability, extubation, and achieving euvolemia on the third day of admission, the patient was started on guideline-directed medical therapies for his heart failure. He remained in normal sinus rhythm without any recurrent episodes of AF.
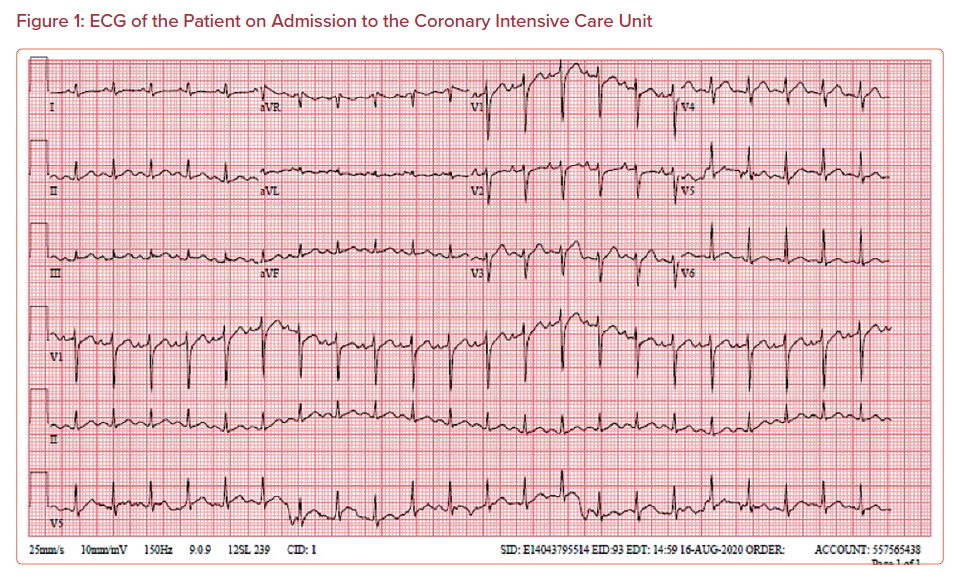
The patient eventually underwent cardiac MRI, which showed a recovery of biventricular function with an ejection fraction of 55% and a minimal non-specific strip of late gadolinium enhancement in the basal septum (Figure 2). Given the absence of a clear culprit explaining this patient’s new systolic dysfunction, the leading hypothesis was that one of his antipsychotic medications, specifically clozapine, caused drug-induced cardiomyopathy. The patient was discharged on day 8 of hospitalization on guideline-directed medical therapies. One month after discharge, the patient was evaluated in the outpatient clinic and was found to be euvolemic on examination with New York Heart Association Class II symptoms.
Discussion
We present a case of a young patient with a history of paranoid schizophrenia who was diagnosed with drug-associated cardiomyopathy. In this case, the patient presented with cardiogenic shock one month following a recent hospitalization during which clozapine was started, an antipsychotic drug associated with cardiotoxicity. The diagnosis of drug-induced non-ischemic cardiomyopathy is usually an exclusion factor. The patient responded well to the stabilizing measure using invasive cardiac monitoring to aid in the diagnosis and management of his shock, with full recovery of his systolic function in the absence of the presumed offending drug.
Drug-induced cardiomyopathy is a less documented and researched complication of clozapine compared with clozapine-associated myocarditis.4 The presentation of clozapine-associated cardiomyopathy resembles that of acute heart failure, but a significant proportion (40–83%) of patients may be asymptomatic, which may underestimate the actual prevalence of this complication and complicate its diagnosis.5 The patient’s invasive hemodynamic measurements, along with his initial presentation and laboratory workup, were consistent with Society for Cardiovascular Angiography & Interventions stage C cardiogenic shock.6 It is critical to evaluate these patients for possible acute coronary syndromes, which may require urgent revascularization. Furthermore, a detailed history may help in distinguishing between a scenario of acute heart failure or an acute decompensation of a chronic condition. Here, a left heart catheterization excluded a potential ischemic etiology, making a non-ischemic cause more likely to have led to this patient’s acute presentation. Non-ischemic etiologies of acute heart failure include: uncontrolled hypertension, arrhythmias, myocarditis, medications (e.g. non-steroidal antibiotics, antineoplastic agents), valvular heart disease, stress-induced (takotsubo), and cardiotoxicity. The diagnostic tools used during this patient’s hospitalization and his history of initiating a new medication with known cardiotoxicity raised the suspicion of a drug-induced cardiomyopathy progressing into cardiogenic shock.
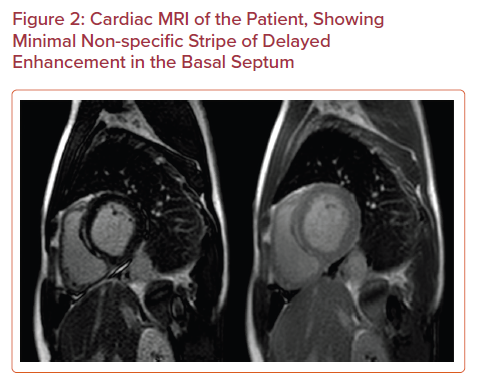
We summarize the cardiotoxicities associated with commonly used antipsychotic medications in Table 1. Clozapine is known to be associated with drug-induced cardiomyopathy; it is a Food and Drug Administration-approved atypical antipsychotic medication frequently prescribed for treatment-resistant schizophrenia.7 It is the only antipsychotic agent that has been proven to significantly reduce suicide among this patient population and does so by up to sixfold.8 While clozapine has been associated with significant cardiotoxicity, a longitudinal study of 62,250 patients followed over a period of 20 years showed that those on clozapine have the lowest overall mortality and fewest deaths due to ischemic heart disease relative to the eight most used antipsychotic agents.9 Fortunately, the prevalence of clozapine-associated cardiomyopathy is very low (0.01–0.19%); however, its incidence is associated with significant mortality (10–46%).10 While clozapine is more recognized as being associated with severe neutropenia and agranulocytosis, its cardiotoxic profile remains less recognized, mainly due to its challenging and highly variable clinical presentation, along with its very low prevalence.4 Nevertheless, there have been three major cardiotoxic adverse complications associated with clozapine reported in the literature: clozapine-associated myocarditis, subclinical clozapine-associated cardiotoxicity, and clozapine-associated cardiomyopathy.11
In a review of 26 case reports of clozapine-induced cardiomyopathy, the most common presenting symptom was shortness of breath (60%) followed by palpitations (36%).12 The mean age of these patients was 33.5 years and the average time for symptoms to develop was 14.4 months, with cases occurring as early as 3 weeks after drug initiation. The echocardiographic findings were consistent among patients with clozapine-associated cardiomyopathy showing reduced ejection fraction with global systolic dysfunction. Interestingly, clozapine-associated cardiomyopathy does not seem to be dose-dependent, as patients were on several dosages prior to presentation (125–700 mg).
While the exact mechanism underlying clozapine-induced cardiomyopathy is still not evident, investigators have posed several hypotheses. Some studies suggested that a defect in CYP1A2 and CYP3A4 enzymes, which regulate clozapine metabolism, may contribute to this complication.13 Others have reported a possible association between selenium depletion and clozapine-induced cardiomyopathy.14 Interestingly, higher rates of clozapine-associated cardiomyopathy and myocarditis have been documented in New Zealand and Australia, raising the possibility of ozone-mediated M2 receptor blockade and cholinergic receptor dysfunction in triggering cardiomyopathy.15
Other studies investigating the mechanisms underlying clozapine-induced myocarditis have shown that this drug may initiate a hypercatecholaminergic state, and the use of β-adrenergic blocking agents may attenuate the incidence and severity of clozapine-induced myocarditis.16 Earlier studies have associated clozapine cardiotoxicity with an immunoglobulin E-mediated hypersensitivity reaction supported by histological findings of eosinophilic inclusions and serological findings of eosinophilia.17
Because clozapine is a highly effective medication in treating schizophrenia, close monitoring and vigilance is critical to prevent deleterious complications associated with drug cardiotoxicity. To date, there is no evidence or consensus supporting pre-emptive screening.7 According to the American Psychiatric Association, whenever clozapine-induced myocarditis or cardiomyopathy is suspected, a cardiology consult is warranted.7 One strategy that may decrease the likelihood of cardiotoxicity is slow and gradual titration of the medication with individualized dosing based on therapeutic drug monitoring and factors known to impact CYP1A2 metabolic activity, including ethnicity/ancestry, smoking status, and sex.7,18,19 Rapid clozapine titration, which occurred in this case, can result in inflammatory release of cytokines that reduce CYP1A2 activity, leading to a positive feedback mechanism of further increase in serum clozapine levels and additional inflammation.20 One titration approach for an individual with presumed average metabolism (i.e. non-smoking male with European or Western Asian ancestry) could be to initiate clozapine at 12.5 or 25 mg in the evening of the first day. If tolerated, the dose is increased to 25 mg on the second day. Daily dosage could be increased gradually by 25 mg to reach a dose of 100 mg/day by the end of first week, and 200 mg by the end of the second week.20
Ronaldson et al. have developed a cardiac monitoring protocol extending over 4 weeks for clozapine cardiotoxicity.21 The authors recommend baseline troponin, C-reactive protein (CRP), and echocardiography upon drug initiation. This is followed by daily symptom assessment and a hemodynamic assessment on every other day. A biochemical assessment of CRP and troponin levels is warranted every 7 days. The authors recommend clozapine caseation if troponin rises above twice the upper normal limit or if CRP levels exceeds 100 mg/l. However, they recommend continuing clozapine therapy with more frequent monitoring to avoid interruption of clozapine titration and limiting association inflammatory response: if symptoms arise, pulse rate exceeds 120 BPM or is elevated from baseline by 30 BPM, CRP levels 50–100 mg/l, or if troponin rises by less than double the normal upper limit. Another critical factor to consider when initiating clozapine is drug–drug interactions. Ronaldson et al. recommend minimizing polypharmacy while titrating clozapine, as several drugs may exacerbate clozapine’s cardiotoxicity, including sodium valproate, lamotrigine, fluvoxamine, and lithium.21 Nevertheless, it is important to educate all patients and their caregivers about the early signs of clozapine cardiotoxicity.
The management of clozapine-associated cardiomyopathy includes clozapine cessation and heart failure guideline-directed medical therapy.7 Clozapine suspension along with conventional heart failure management consisting of angiotensin-converting enzyme inhibitors, β-blockers, and diuretics have led to a significant improvement in left ventricular function.22 Decisions regarding resuming clozapine therapy are highly individualized, and should consider weighing in the risks and benefits of treatment.7 Whenever clozapine is rechallenged, very close monitoring and frequent echocardiography may be warranted to prevent subsequent cardiotoxicity.23
Conclusion
We treated a case of clozapine-associated cardiomyopathy presenting in cardiogenic shock. Drug-induced cardiomyopathy is a common yet underrecognized etiology of non-ischemic cardiomyopathy. Therefore, clozapine-associated cardiomyopathy should be considered as a differential diagnosis in schizophrenic patients presenting with signs of acute heart failure.
Click here to view Supplementary Video.
Clinical Perspective
- High clinical suspicion is needed to diagnose drug-induced cardiomyopathy when investigating the cause of non-ischemic cardiomyopathy.
- Clozapine is an effective medication for treatment-resistant schizophrenia.
- Clozapine is reported to be associated with drug-induced cardiomyopathy and is more common with rapid drug titration. Clozapine is more commonly associated with myocarditis.
- Close monitoring and vigilance is critical to prevent cardiac complications associated with initiating clozapine.