







One of the fundamental principles of evidence-based medicine is that clinical care should be based on data derived from appropriately designed trials, registries, and observational data from patients. The best available evidence is then used to develop guidelines for clinical care, assess quality, measure performance, and improve patient outcomes. The highest level of evidence in clinical medicine, also known as Level of Evidence A, is derived from multiple prospective randomized clinical trials (RCTs) or from meta-analysis. Data derived from a single randomized prospective trial, nonrandomized studies, or populations evaluated in registries are considered to be Level of Evidence B. The least robust types of evidence in clinical medicine are consensus opinion of experts, case studies, and standard of care opinion, which are considered to be Level of Evidence C. These levels of evidence provide clinicians with an estimation of certainty regarding treatment effect. It is evident that Level of Evidence A has the highest and C has the lowest certainty. While the best available evidence is always used by professional societies to guide clinical care, this evidence meets the standards of Level A in only approximately 30 % of guideline recommendations.
While RCTs are regarded as the ‘gold standard’ of evidence-based medicine, they are not feasible for many conditions due to small patient populations, ethical considerations, lack of feasibility, or insufficient funding. Increasingly, registries have played a major role in bridging gaps in knowledge when data from RCTs are not available. By definition, registries collect data regarding diagnosis, treatment, and outcomes based on physician’s judgment without any mandated interventions. While there are many limitations to registries, the collective data can be used to assess many dimensions of diagnosis, natural history, risk stratification, and treatment. More recently, registries have become an essential research tool in assessing genotype–phenotype relationships, with profound implications for family members of probands.
Contemporary disease registries in cardiovascular medicine have demonstrated the capacity to favorably impact clinical management of uncommon conditions, particularly arrhythmogenic cardiovascular conditions.1 The patient populations in disease registries represent unbiased samples that most resemble the true clinical population, rather than patients selected based on the restrictive inclusion and exclusion criteria of clinical trials.1 Registries, therefore, contribute valuable insight into large ‘real-world’ populations over time.
Among the conditions for which only registry data are available to guide clinical care are multiple cardiovascular conditions manifesting with cardiac arrhythmias. Despite the absence of RCTs for these arrhythmogenic disease entities, knowledge has been advanced sufficiently to result in meaningful improvements in patient outcomes based solely on registry observations. The prototype registry was established for patients with the congenital long QT syndrome (LQTS).2,3 This syndrome is characterized by a long QT interval (corrected QT interval [QTc] >440 msec), stress-induced syncope, and the occurrence of life-threatening tachyarrhythmias.2,3 Over three and a half decades, this registry has served as the foundation for meaningful advances in diagnosis, risk stratification, treatment, and useful knowledge related to genotype–phenotype relationships for patients with this condition.
Other cardiovascular syndromes commonly manifesting with arrhythmias include arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/C), Brugada syndrome, hypertrophic cardiomyopathy (HCM), and cardiac sarcoidosis (CS). The focus of this review is advances in knowledge provided by contemporary registries related to these cardiovascular conditions commonly manifesting with cardiac arrhythmias. Currently, clinical diagnosis and evidence-based treatment of these conditions result in an excellent long-term prognosis. By contrast, failure to diagnose and appropriately treat patients with these conditions can result in fatal outcomes. In this respect, informed diagnosis and treatment are considered to be clinical imperatives.
Based on the relatively small patient populations and the multiple considerations noted above, RCTs have not been conducted for these conditions. A systematic review of the literature, using PubMed, Ovid, and Google Scholar, was conducted related to ARVD/C, Brugada Syndrome, HCM, LQTS, and CS for multicenter, prospective registries. To provide the best available contemporary data, registries were included if they enrolled at least 50 patients, had published results after 2010, and had a follow-up of at least 2 years. After a systematic review of all registries, the knowledge gained was classified into multiple dimensions including: defining diagnostic criteria, characterizing the natural history, risk stratifying, therapy, and genotypic–phenotypic relationships for the cardiovascular conditions.
Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy
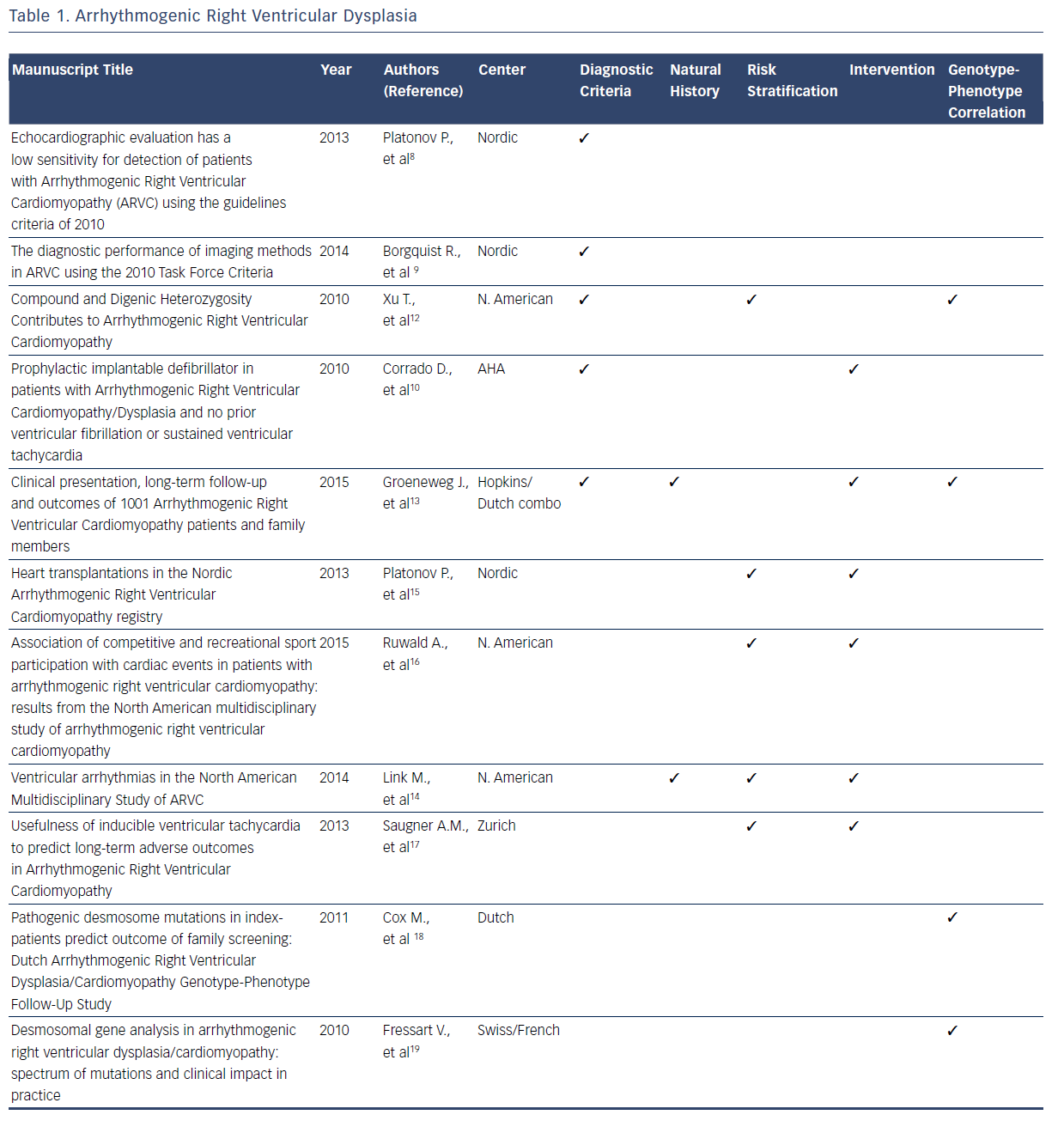
ARVD/C is a rare, inherited disorder characterized by patchy replacement of ventricular myocardium by fibrofatty tissue, commonly manifesting with ventricular arrhythmias and sudden death.4–19 After the original description in 1977, collective data from registries served as the foundation for the original diagnostic criteria for ARVD/C developed in 1994.4 Multiple registries were subsequently used to refine the diagnostic criteria in 2010.5,7 These new diagnostic criteria have a higher sensitivity and specificity for the condition compared with the 1994 criteria. With the 2010 ARVD/C diagnostic criteria, national and international databases have provided insights into the advantages and limitations of endocardial voltage mapping cardiac imaging modalities, genetic testing, and family history that will be incorporated in future revisions of the diagnostic criteria.8–13Table 1 summarizes the contributions of registries included in this systematic review to the current diagnostic criteria and knowledge related to the natural history, risk stratification, intervention, and genotype–phenotype relationship for ARVD/C.
Beyond the refinement of diagnostic criteria, registries have also elucidated many aspects of the natural history of patients with ARVD/C.4–19 Among the most valuable insights gained from registries is that this condition is associated with a high frequency of lifethreatening ventricular arrhythmias. A considerable amount of the registry information related to ARVD/C addresses the clinical course of patients with implantable cardioverter defibrillators (ICDs) versus those without ICDs.12–14 Valuable insights have been gained into risk stratification for ventricular arrhythmias.12–17 In particular, registries have confirmed that patients who participate in competitive sport have earlier onset of symptoms and an increased risk of ventricular arrhythmia than those who participated in recreational sport or who were inactive.12–17 Spontaneous or induced ventricular arrhythmias and younger age at presentation have also been established as risk factors for future ventricular arrhythmiasbased analyses of multiple registries.14,17 Additionally, it has now been established that the presence of one plakophilin-2 (PKP2) variant or other desmosome-encoding gene variant results in earlier onset and more severe ARVD/C than those who are heterozygous.12
Management and therapeutic interventions for patients with ARVD/C have been developed based on observations provided by registries. The consistent observation in registries that competitive athletics results in disease progression commonly manifesting with ventricular arrhythmias, has resulted in the recommendation for restriction of competitive athletics.11,16,17 A recent analysis of a North American registry suggests that recreational sports participation may be as safe as no athletic participation.16 The frequency with which patients have episodes of ventricular tachycardia requiring intervention by an ICD has been defined by registries.10,13,14,16,17 The many clinical similarities of isolated and familial ARVD/C has been elucidated from one observational registry.13 Finally, registries have consistently showed that the need for patients with ARVD/C to advance to heart transplant is rare.10,13–17
Another consistent finding among contemporary registries is that pathogenic desmosomal gene mutations, mainly truncating PKP2 mutations, correlate with the clinical manifestation of ARVD/C.11–13,18,19 This pathogenic gene is associated with a sixfold risk of ARVD/C diagnosis in mutation-carrying relatives when compared with relatives of patients without mutations.18,19 However, the North American registry data suggest that harboring one PKP2 mutation may not be sufficient to determine overt clinical disease.7,11 Compound or digenic heterozygosity, acquired disruption of proteins, or environmental factors were required in 42 % of cases for overt clinical phenotype.11 A Swiss/French registry identified 41 disease-causing mutations in patients with ARVD/C, indicating that there is a large spectrum of mutations with multiple mechanisms including missense, splicing, frameshift, and deletions mutations.19 It is evident that the knowledge regarding the genotype– phenotype relationships for ARVD/C is incomplete. Currently, a large National Institutes of Health-funded registry is enrolling patients with the objective of elucidating these complex relationships.
Brugada Syndrome
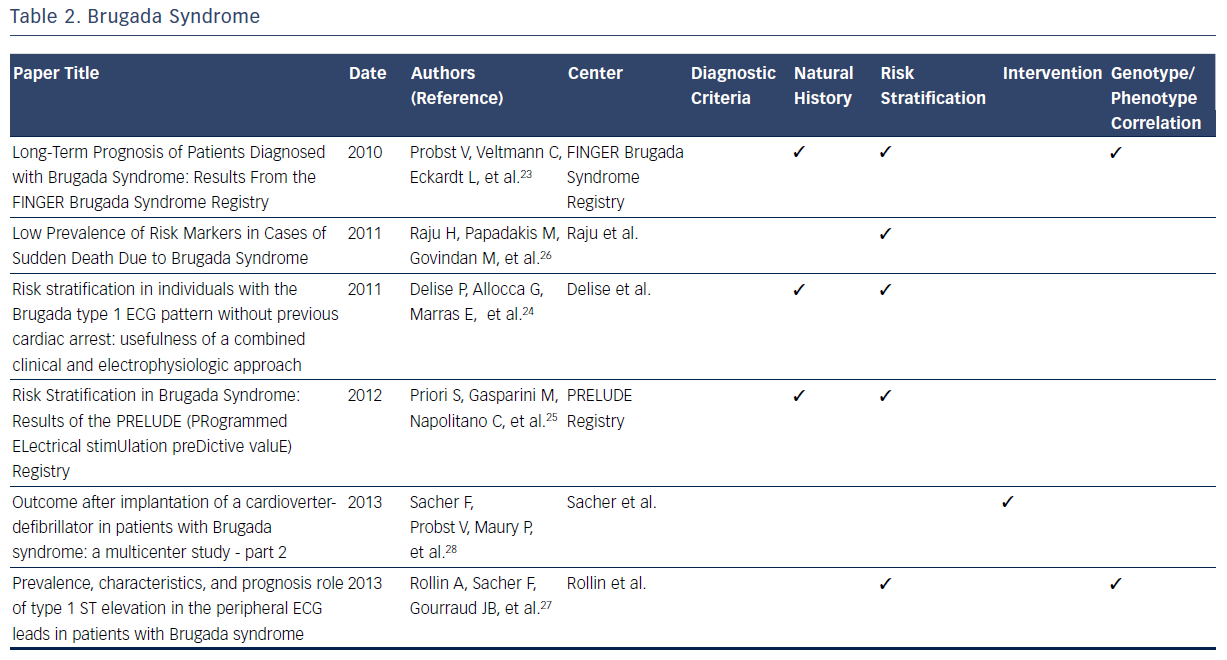
The Brugada syndrome is characterized by ST-segment elevation in the right precordial leads, episodes of ventricular fibrillation, and sudden cardiac death (SCD).20–28 Observations from registries allowed development of formal criteria in 2005.20 Since then, registries have independently reported on many aspects of the natural history of those identified with symptomatic and asymptomatic Brugada syndrome.20–28
One large contemporary registry has allowed elucidation of the annual cardiac event rates for patients with a previous episode of aborted SCD (7.7 %).23 By contrast, those presenting with syncope have a considerably lower annual incidence of life-threatening ventricular arrhythmia (1.9 %).23 Finally, asymptomatic patients have been noted to have a low annual rate of major arrhythmic events (0.5 %).23 Two other large prospective registries have confirmed these important findings regarding risk stratification in patients with Brugada syndrome.23–25 These consistent findings among multiple registries of the relatively low annual sudden death rate in those without prior cardiac arrest have helped clinicians make decisions regarding the risks and benefits of ICD placement in this patient population.23–25
Data from one registry suggest that the majority (72 %) of SCDs in patients with Brugada syndrome are in low-risk, asymptomatic patients.26 This observation highlights the challenge of predicting and preventing sudden death in low-risk patient populations. In addition to aborted SCD and syncope, a spontaneous type 1 ECG has been noted to be an independent predictor of arrhythmic events.23 In addition to the France, Italy, Netherlands, Germany FINGER registry predictors, the Programmed Electrical Stimulation Predictive Value (PRELUDE) registry identifies ventricular effective refractory period of <200 msec and QRS fragmentation as independent risk indicators able to identify patients at high risk for ICD implantation.25 In contrast to previous findings, the FINGER registry indicates that VT/VF inducibility during programmed electrical stimulation is found to have no predictive value in identifying high-risk patients.25 Other factors, including family history of SCD, gender, inducibility of ventricular tachyarrhythmias during electrophysiological study and the presence of an SCN5A gene mutation, have been noted to have limited predictive value.23 These findings directly impacted on therapeutic recommendations identified in the 2005 Brugada Syndrome: Report of the Second Consensus Conference.20
A consistent observation from Brugada syndrome registries is that ICD implantation is the only effective treatment for prevention of SCD.20 Appropriate shock rates during follow-up after ICD implantation are highest for patients implanted with ICDs due to a previous aborted sudden cardiac arrest and lower for those with syncope.28 The limited available data regarding ICD shocks in asymptomatic patients demonstrates a low frequency of appropriate shocks.28 The appropriate shock rates are greatest in symptomatic patients as compared with asymptomatic patients.28 While optimal ICD programming and follow-up increased appropriate shock rates, lead failure proved to be an issue throughout patient follow-up (29 % at 10 years).28
One large registry demonstrated that the presence of a mutation in the SCN5A gene lacks predictive value for arrhythmic events.23 Analysis from risk stratification data from another indicates that type 1 ST elevation in peripheral ECG leads are significantly more likely to contain SCN5A mutations, and express a more severe Brugada syndrome phenotype with a higher risk of arrhythmic events.27 Overall, contemporary registries consistently report a low prevalence of SCN5A mutations among the studied cohorts, suggesting a lack of diagnostic contribution from genetic testing.20–28Table 2 summarizes the contributions of registries to many dimensions of current knowledge related to the Brugada syndrome.
Hypertrophic Cardiomyopathy
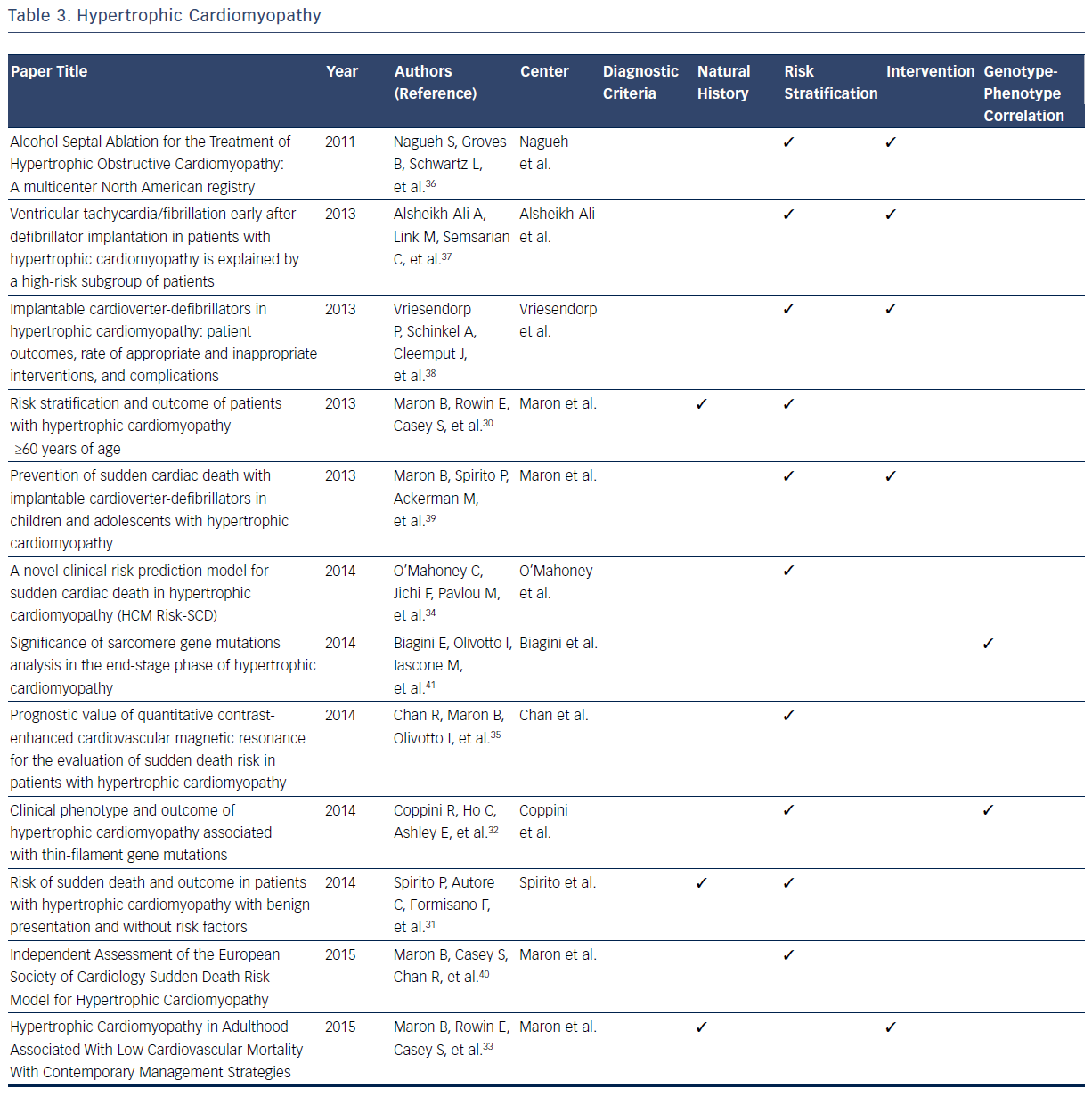
Hypertrophic cardiomyopathy (HCM) is a disease caused by a mutation in the sarcomeric genes, which can present with SCD, ventricular arrhythmias, or, more insidiously, with heart failure.29–40 With a high prevalence of this condition in the general population, estimated at 1 in 500 individuals, large databases have defined many dimensions of the condition.29–41 Publications based on data from these registries have been the foundation of defining diagnostic classification for the condition.29
In addition, all aspects of the natural history of HCM have been derived from registries. It is now known that 88 % of patients with HCM present at age ≥60 years, primarily with absent or mild HCM-related symptoms, and typically die from non-HCM related causes.29,32 Maron et al identified 3.7 % of patients who suffered HCM-related mortality (progressive heart failure or transplant, embolic stroke, or arrhythmia-related death).30 Based on registry data it has become evident that asymptomatic patients or those with mild symptoms have a relatively favorable course with only 5.4 % of patients suffer HCM-related sudden death.30 SCD is found to be independently and inversely related to age, whereas heart failure and stroke are directly related to age.31 One large contemporary registry suggests that high-risk patients who survive life-threatening events (5.6 %) commonly survive in the setting of ICD intervention or heart transplant, thus implying that modern advances may improve mortality rates in this subgroup.33
Further registry data suggest that all-cause mortality rates are increased in patients with HCM aged ≥60 years when compared with an agematched population, predominantly as a result of non-HCM–related diseases.33 Spirito et al found SCD risk was not entirely negligible (event rate of 0.6 % per year) in patients without conventional risk factors and with absent to mild symptoms.31 A recent multicenter cohort study found that age, maximal left ventricular (LV) wall thickness, left atrial diameter, LV outflow tract gradient, family history of SCD/non-sustained ventricular tachycardia, and unexplained syncope are associated with appropriate ICD shock or SCD in patients with HCM.34 These predictors are used to model the possibility of SCD after 5 years, and it was concluded that most patient with HCM with SCD or appropriate ICD interventions are misclassified with low-risk scores.34 Of the 1629 patients examined, 35 experienced SCD, of which only four have a high predictive risk score that is consistent with a recommendation of ICD implantation. These conclusions indicate the need for continued assessment of accurate risk stratification in this particular population.33 A study examining cardiovascular MRI found that the extent of late gadolinium enhancement is associated with an increased risk of SCD.35
Clinical outcome following septal alcohol ablation demonstrates survival rate estimates at 1, 5, and 9 years being 97 %, 86 %, and 74 %, respectively.36 Predictors of mortality include low baseline LV ejection fraction, lower number of septal artery ethanol injection, higher number of ablation procedures per patient, high post-ablation septal wall thickness, and requirement of beta-blockade post-procedure.36 Other studies have examined the effectiveness of ICD implantation in patients with HCM. It has been noted that 22 % of patients with ICD implantation receive at least one subsequent ICD shock.37 The risk of discharge is highest in the first year post-transplant (10.8 % per person-year), and more frequent in patients who receive ICD implantation as a secondary prevention measure.37 Similarly, the frequency of appropriate ICD shock is higher in both patients receiving implantation for secondary prevention and in male patients. Inappropriate ICD interventions and device-related complications occur in 3.7 % and 3.6 % of patients, respectively.38 Registries also give important insights into outcomes of pediatric patients with ICD implantation.40 Appropriate interventions occur in 19 %, with mean time from implant to first appropriate shock being 2.9±2.7 years.40 Extreme LV hypertrophy is considered the most common risk factor for patients receiving primary prevention ICD therapy.40
A study comparing a cohort of patients with end-stage HCM (ES-HCM) with a reference cohort of HCM patients with normal LV ejection fraction concluded that ES-HCM is associated with various genetic substrates indistinguishable from patients in the reference cohort, with the exception of a larger number of complex genotypes of sarcomere genes.41 In addition data from this registry indicate that thin-filament mutations are associated with an increased likelihood of advanced LV dysfunction and heart failure when compared with thick-filament mutations. The risk of arrhythmia in these two subsets is comparable.32Table 3 provides an overview of areas in which registries have expanded current knowledge related to the HCM.
Long QT Syndrome
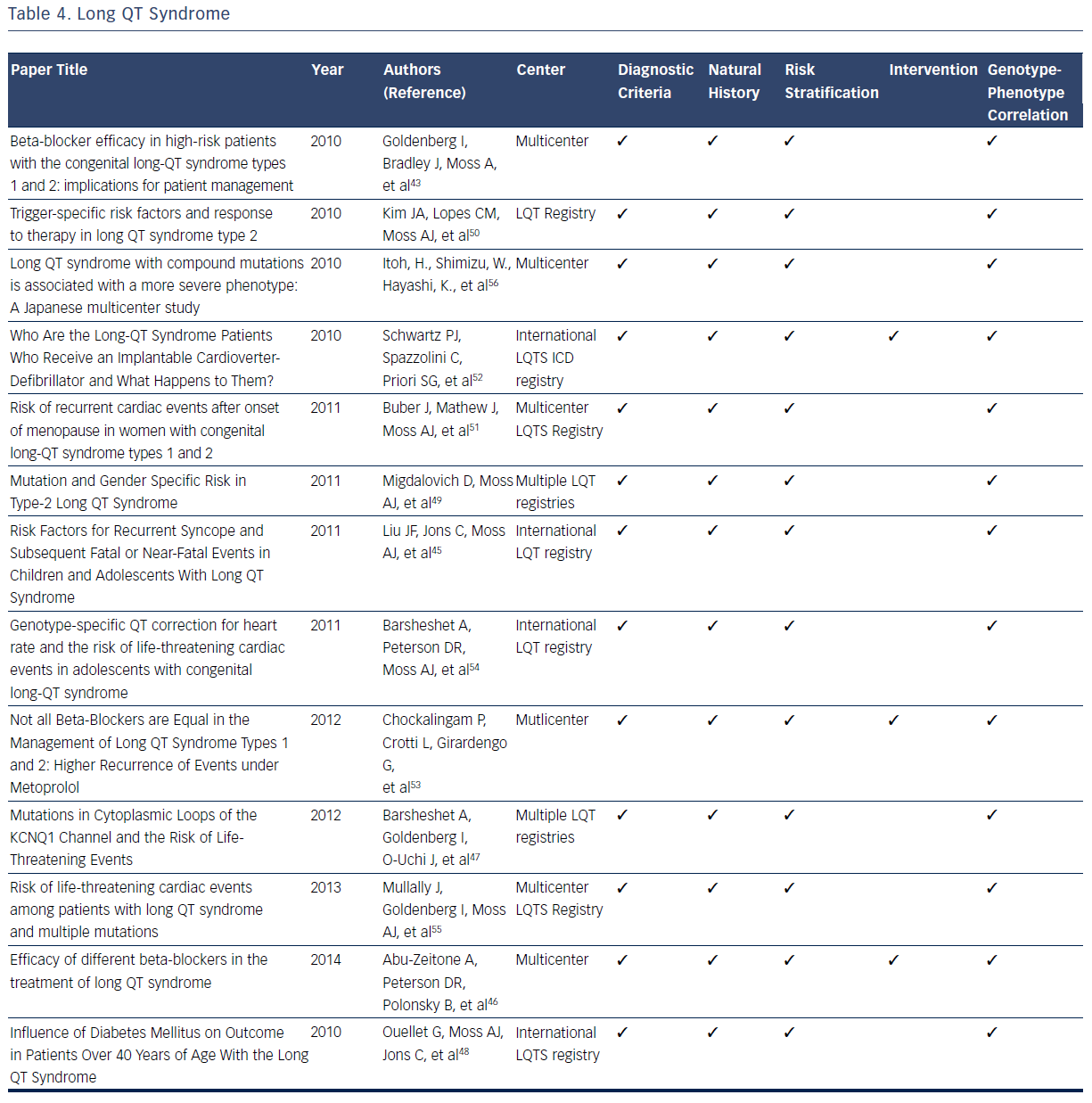
The LQTS is characterized by a long QT interval (QTc >440 msec), stressinduced syncope, and the occurrence of a unique form of ventricular tachycardia know as Torsades des Pointes.2,3 LQTS is estimated to have a prevalence of 1 in 3,000 to 1 in 5,000.42–56 The original International LQTS Registry and the many subsequent registries have filled many knowledge gaps related to diagnosis, risk stratification, treatment, and genotype–phenotype relationships for patients with this condition.42–56 On the basis of registry data, formal diagnostic criteria for the condition have been developed.42–56 For diagnostic ECG, the QT interval is measured manually from the beginning of the QRS complex to the end of the T wave. QTc is obtained using Bazett’s, Fridericia, or Framingham correction formula.2,3,42–44
LQT registries have provided many clinical insights related to risk stratification for syncope and sudden death that profoundly influence clinical decisions. Multiple studies identify prolonged QTc duration (≥500 msec) and 100-msec increments in the absolute QT interval as electrocardiographic markers of increased risk for syncopal episodes and life-threatening cardiac events.44,45,47 Other risk factors for syncope, prior SCD, or aborted cardiac attacks include ≥1 episodes of syncope and presence of LQTS genotypes.44,45 There is no observed risk increase of syncope or ventricular arrhythmias in patients with LQTS taking oral contraceptives.46 In addition, development of diabetes in adult patients with LQTS is not associated with an increased risk of first cardiac events dominated by syncope.48
These clinical and electrocardiographic criteria have been supplemented and refined by genetic confirmation of LQT gene mutations. Studies from the International LQTS registry indicate that a greater risk for cardiac events exists for male carriers of the LQT1 gene and female carriers of the LQT2 gene.43,44 For patients specifically diagnosed with type 2 LQTS (LQTS2), female gender has been demonstrated to be a powerful marker of increased risk of life-threatening cardiac events and the first occurrence of trigger cardiac events.49,50 The onset of menopause is associated with a significant increase in the risk of cardiac events in female carriers of LQT2, which suggests careful follow-up and long-term therapy are warranted in this population.51
Multiple registries have demonstrated that syncope and cardiac arrest could be reduced dramatically in patients with LQTS by use of beta-blockers.42–56 Accordingly, beta-blockers have emerged as the initial therapeutic option in patients with LQTS since the original observation of the therapeutic effect of these agents.43,50 Several contemporary studies validate their long-term efficacy in reducing the risk of cardiac events.43,50 Beta-blockers should be routinely administered to all patients at high risk of type 1 (LQT1) and LQT2 as they significantly reduce the risk of life-threatening events in both male carriers of LQT1 and female carriers of LQT2.42 Differential efficacy of betablockers has been suggested for treating LQTS.46,54 Among patients with a prior cardiac event while taking beta-blockers, efficacy for recurrent events differ by drug, with propranolol being the least effective compared with other beta-blockers.46 It has also been consistently observed that patients with recurrent syncope while taking beta-blockers have an increased rate of subsequent cardiac events.44 On the basis of these observations it is currently recommended that ICD therapy be provided for this subset of patients with LQTS.44 In addtion, ICD therapy is recommended for all patients with LQTS who have survived a prior episode of cardiac arest.52
Genotype–phenotype correlations in the LQTS population have been facilitated by the commercial availabity of genetic testing. The robust clincal data available in multple registries have demonstrated that multiple mutations, compound mutations in either single or different genes, are associated with a more severe phenotype and a greater risk of lifethreatening cardiac events.54–56 Additional mutations, including mutation characteristics (transmembrane-missense versus nontransmembrane or nonmissense mutations), pore-loop mutations,49 and the presence of C-loop mutations in LQTS1, have been associated with greater risk of cardiac events.44,47,54Table 4 summarizes the considerable contributions of registries related to the diagnosis, natural history, risk stratification, treatment, and genotype–phenotype relationships for LQTS.
Sarcoidosis
CS affects a small minority of patients with pulmonary or systemic sarcoidosis, but when present is often associated with a spectrum of clinically significant conduction abnormalities and arrhythmias.57–68 Unlike other inherited arrhythmogenic cardiovascular conditions reviewed, sarcoidosis is not clearly identified as an inherited disorder. However, epidemiologic data suggest some genetic predisposition.57,60 It has become evident through registry data that the cardinal manifestations of CS are conduction disturbances and arrhythmias.57–68 Although sarcoidosis is typically a multisystem granulomatous disease with cardiac involvement in <10 % of patients with multisystem disease, isolated CS is a definite clinical entity.57–69 As CS commonly presents with life-threatening heart block, malignant arrhythmias, and congestive heart failure, detection and appropriate treatment are essential to ensure the best possible patient outcomes. Registries have played an essential role in filling the knowledge gaps in many domains related to CS.57–69 A recent consensus statement provides guidance for clinicians on the diagnosis and management of arrhythmias associated with CS including indications for ICDs.57 The current diagnostic criteria, natural history, and risk stratification process for CS are all based on data from multiple registries.57 In the therapeutic domain, pacemakers, ICD implantation, and early implementation of corticosteroid therapy have led to an improvement in the overall prognosis and clinical outcomes of CS based on observations from registries.57–69
Contemporary registries contributed to the recent establishment of CS diagnostic criteria in 2014.57,58 The 2014 criteria place greater emphasis on patients with proven systemic sarcoidosis, which is supported by the findings of Schuller et al.58 Blankstein et al. showed a correlation between 18F-fluorodeoxyglucose (18F-FDG) uptake on positron emission tomography (PET) scans and poor outcomes, thus allowing patients to now be officially diagnosed via PET scan evidence.59 The usefulness of MRI evidence has expanded with respect to CS diagnostic criteria refinement.59,60
Recent registries contribute to the knowledge of risk stratification in CS patients. Two registries have specifically improved the usefulness of MRI and PET imaging. Crawford et al. demonstrated that delayed gadolinium enhancement in the right ventricle is associated with adverse events in patients who have a preserved ejection fraction and no prior history of ventricular tachycardia.63 Furthermore, delayed enhancement is associated with a high risk for recurrent ventricular tachycardia in patients with a history of ventricular tachycardia and enhancement. MRIs with gadolinium also make good negative indicators, as a lack of delayed enhancement is associated with a low risk for ventricular tachycardia.63 Greulich et al. support these results and identify late gadolinium enhancement as the best independent predictor of both adverse and potentially lethal events.64 18F-FDG uptake on PET scans is an indicator of ventricular tachycardia risk and death.59 Takaya et al. concluded that patients with CS who present with high degree atrioventricular (AV) block, as opposed to ventricular tachycardia, are at high risk of fatal cardiac events.65
The primary intervention for CS in the reviewed registries is ICD implantation. Schuller et al. indicate that approximately one-third of patients with CS with implanted ICDs receive at least one appropriate therapy. Based on this finding, Schuller et al. hypothesize that this increased activity could cause scarring, which results in subsequent tachycardia.66 Kron et al. demonstrate that high-degree AV block is a good indicator of the need for a primary prevention ICD.62 Takaya et al. support these findings; patients initially presenting with AV block have similar outcomes to patients presenting with ventricular tachycardia.65 While corticosteroids remain the gold standard for treatment of CS, ICDs are necessary in higher-risk patients.57,60
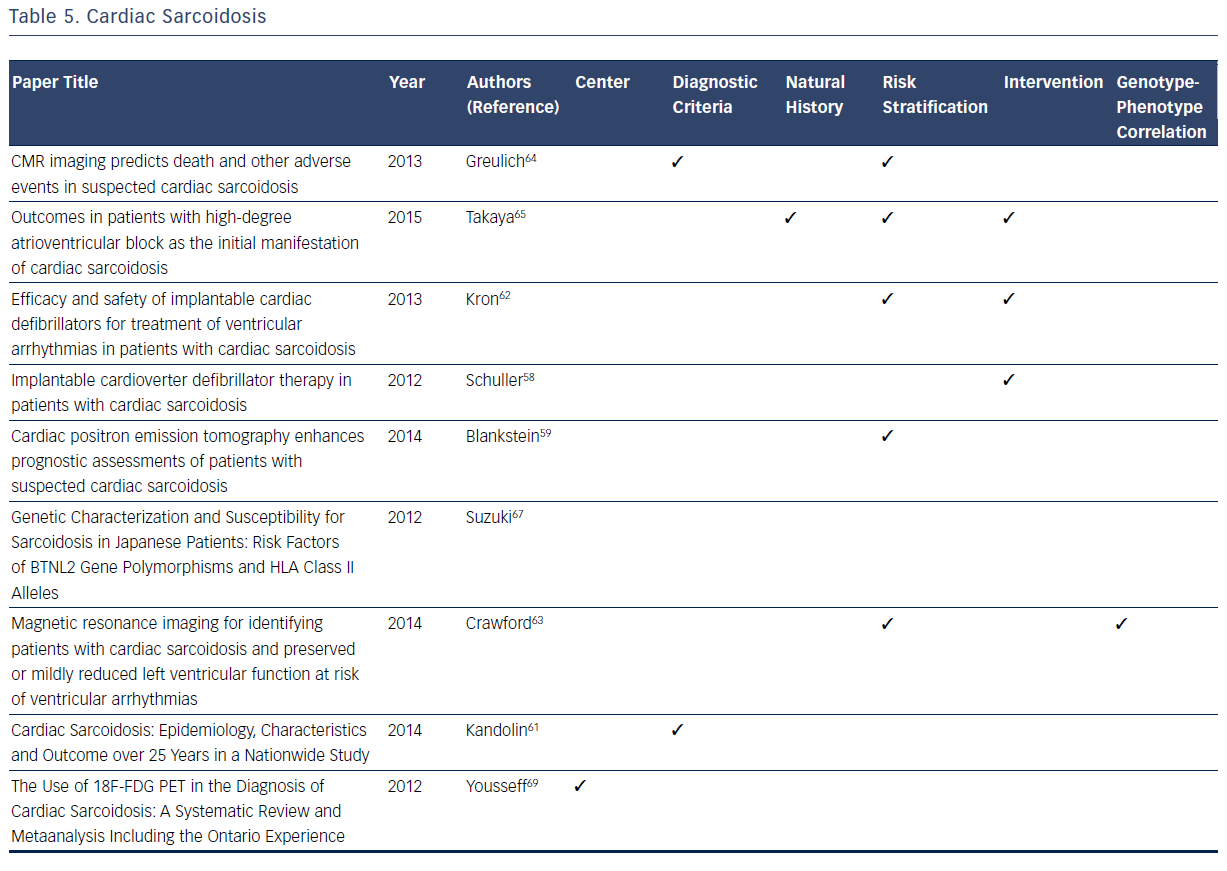
Among the registries reviewed, there is a general lack of information on the genotype–phenotype correlation with CS, with the exception of the research conducted by Suzuki et al.67 This registry confirms that the HLA-DRB1 allele is a major contributing factor in the development of sarcoidosis. However, there is no specific phenotypic correlation with this allele or any other allele with respect to CS symptoms or outcomes.67Table 5 summarizes the considerable contributions of registries to current knowledge related to CS.
Limitations
There are limitations to the methods used in this review. Inclusion of registries published since 2010 is one limitation. Our focus was on new data contributed by contemporary registries rather than on a comprehensive historical review. With this approach, it is possible that unique information published prior to 2010 was not included. Another limitation of the methodology we used is the lack of systematic assessment of registry data quality as part of the selection process for this review. As noted above, registries were included if they enrolled at least 50 patients, had published results after 2010, and had a follow-up of at least 2 years. A limitation of the published data and our review is the inability to assess for enrollment selection bias and generalizability. However, as noted above, the patient populations in disease registries typically represent unbiased samples that most resemble the true clinical population, rather than patients selected based on the restrictive inclusion and exclusion criteria of clinical trials.
Conclusion
Cardiac registries have filled many gaps in knowledge related to arrhythmogenic cardiovascular conditions. When clinical trials are feasible, they demonstrate what can be done. Guidelines inform clinicians what should be done, registries inform clinicians regarding what is actually done. In the absence of clinical trials, registries represent the best available evidence to inform clinicians regarding what can be done. Registries are also used as the best available evidence for guidelines. Thereby informing clinicians what should be done. Despite the less robust level of evidence available in registries when compared with clinical trials, they have contributed clinically useful information related to arrhythmogenic cardiovascular conditions. The more mature registries, such as the LQTS registry, currently provide clinically useful genotype information that has a direct influence on therapy and genetic screening of relatives. It is evident that registries have had a major role in bridging current knowledge gaps related to diagnosis, natural history, risk stratification, treatment, and genetics of arrhythmogenic cardiovascular conditions. Registries also represent the best available evidence for future guidelines, quality metrics, performance measures, and improved patient outcomes.